|
|
 |
|
|
|
Wolfgang Marks: Die Formatierte DNA
|
|
|
Das REMA-Gen-System oder wie Remodeling-Maschinen generiert werden
Rema-Gene kodieren Remodeling-Faktoren.
Die von mir also REMA-Gene genannten DNA-Sequenzen sind vermutlich an das Vorhandensein von repetitiven LINE-Elementen gekoppelt, die sich im Verlauf der Evolution über das ganze Genom ausgebreitet haben. Es gibt deshalb auch eine sehr große Zahl dieser Gene allein in der von mir näher untersuchten Chromatin-Domäne 2 in der Transkriptionsgruppe 2 des Bandes 15q22.275 befinden sich beide reverse Stränge zusammengerechnet - vermutlich 60 REMA-Gene. Darüberhinaus habe ich in der Domäne wiederum den plus- und den minus-Strang zusammengerechnet - eine grosse Zahl von Genen identifiziert, die für RNAs kodieren. Dieser großen Zahl von Protein- und RNA-kodierenden Genen auf dem plus-, dem plus-reverse, dem minus- und dem minus-reverse-Strang steht eine entsprechend große Zahl von Promotoren und SMARs gegenüber.
Die erwähnten Promotoren liegen aber nicht wahl- oder regellos in der DNA verstreut, sondern sind über definierte Nukleosomengrössen zu Informationslayern, zu Netzwerken zusammengefasst, die einer gemeinsamen Steuerung durch distinkte, einheitliche Nukleosomengrössen und Formatierungshormone unterliegen.
|
|
|
|
|
|
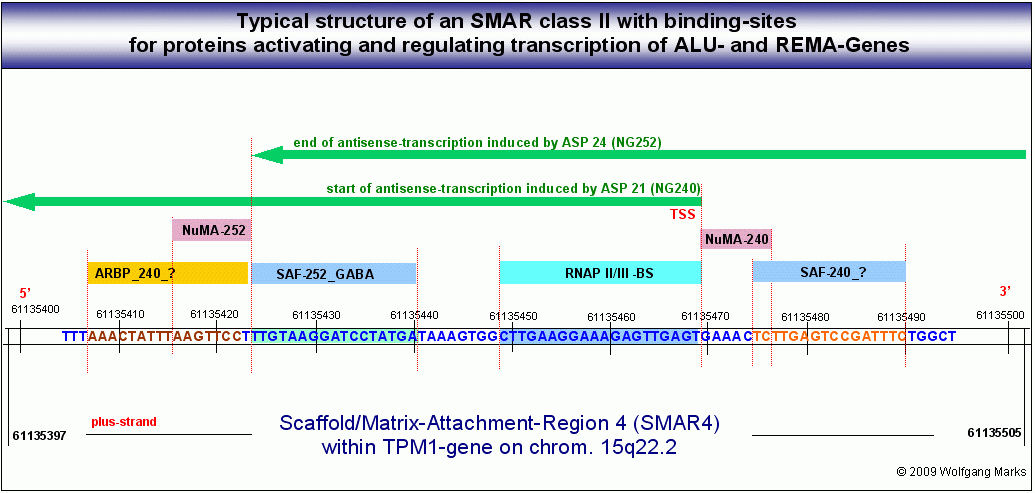
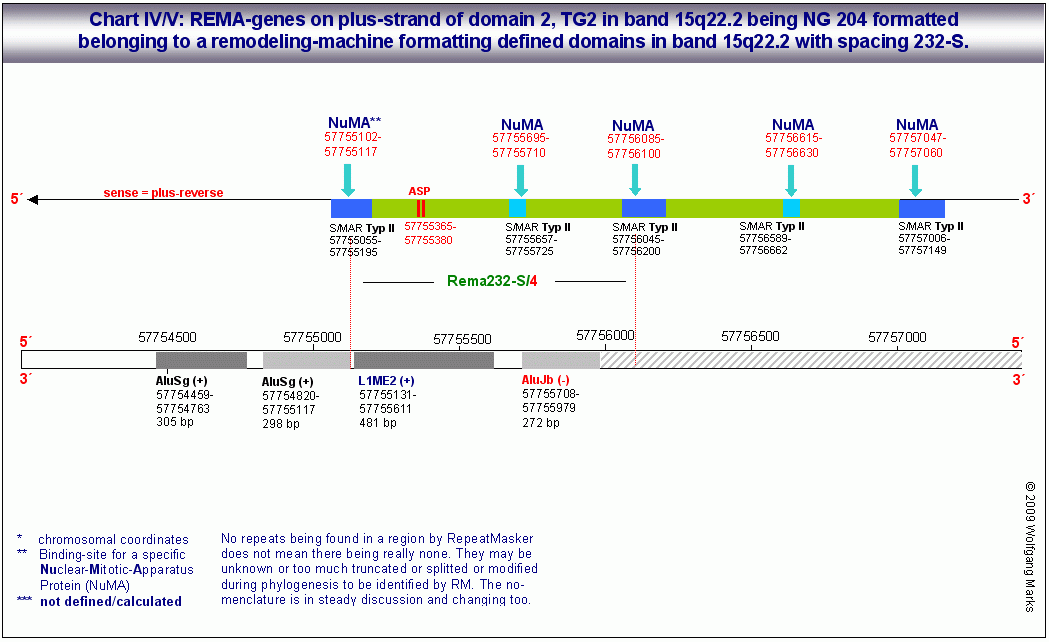
Die zuvor angesprochene Domäne 2 wird durch insgesamt 91 SMARs der Klasse II76 in Transkriptionseinheiten eingeteilt, die sich wie auf der Abbildung rechts oben gezeigt, zum Teil überlappen. Sie enthalten zellspezifische, mit einem definierten Formatierungshormon korrelierte Bindestellen für die zuvor erwähnten NuMA-Proteine und andere Polypeptide, die zu den sogenannten Architekturproteinen gerechnet werden.
Wenn man beliebige, durch SMARs der Klasse II begrenzte, extra- aber auch intragenische DNA-Abschnitte der zuvor genannten Domäne mit Hilfe des ORF-Finder des NCBI (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) auf das Vorhandensein von ORFs untersucht, wird man feststellen, dass sich in jedem DNA-Strang abhängig vom Leseraster, der Transkriptionsrichtung und von der Definition der Mindestgrösse der zu suchenden open reading frames (ORFs) bis zu 10 und mehr solcher ORFs definieren lassen. Die auf diese Weise gefundenen ORFs entsprechen im Prinzip den Exons von normalen polycistronischen Genen und kodieren nach meinen Analysen per se oder gespleisst für kleinere und größere Proteine zwischen ca. 20 und 300 Aminosäuren.
Ich werde nun erläutern, warum ich die auf dem plus-reverse und auf dem minus-reverse (also in antisense-Richtung) kodierten Proteine für die lange gesuchten Komponenten (remodeling-Faktoren) der sogenannten remodeling-machines halte für die entscheidenden Bausteine dieser großen, dynamischen Proteinkomplexe also, denen eine entscheidende Rolle bei der Strukturierung und Organisation des Chromatins und damit bei der Regulation der Genexpression zukommt.
Fast alle gefundenen antisense-ORFs weisen (zum Teil mehrere) Spleiss- und Verzweigungsstellen auf es liegt also nahe anzunehmen, dass der überwiegende Teil, wenn nicht alle ORFs nach der Transkription gespleisst werden. Dafür spricht auch, dass sowohl auf dem plus- als auch auf dem minus-Strang Sequenzen existieren, die mit hoher Wahrscheinlichkeit für spezifische UsnRNAs77 kodieren. Die Transkription dieser UsnRNAs wird sehr oft, wenn nicht überwiegend, von Promotoren aktiviert, die eine Transkription sowohl vom plus- in sense- als auch vom minus-Strang in reverse oder antisense-Richtung initiieren können. Die Transkription der UsnRNA muss demnach mit der der proteinkodierenden REMA-Gene gekoppelt sein. Daraus schliesse ich, dass die in den REMA-Gen-Clustern kodierten UsnRNAs für das Spleissen der REMA-Gen-Transkripte benötigt werden.
Auch die Tatsache, dass die von mir identifizierten sense-Promotoren, welche die Transkription von RNAs auf dem plus- oder minus-Strang aktivieren, mit den antisense-Promotoren, welche die Transkription der remodeling-Faktoren auf dem jeweils gleichen Strang in reverse-Richtung induzieren, nach meinen Analysen über ein Netzwerk distinkter Nukleosomengrössen verknüpft sind, spricht dafür, dass es sich um UsnRNAs handelt, die für das Spleissen der plus-reverse- und minus-reverse kodierten remodeling-Faktoren benötigt werden, da ihre Aktivierung während der Mitose78 sonst wenig Sinn machen würde.
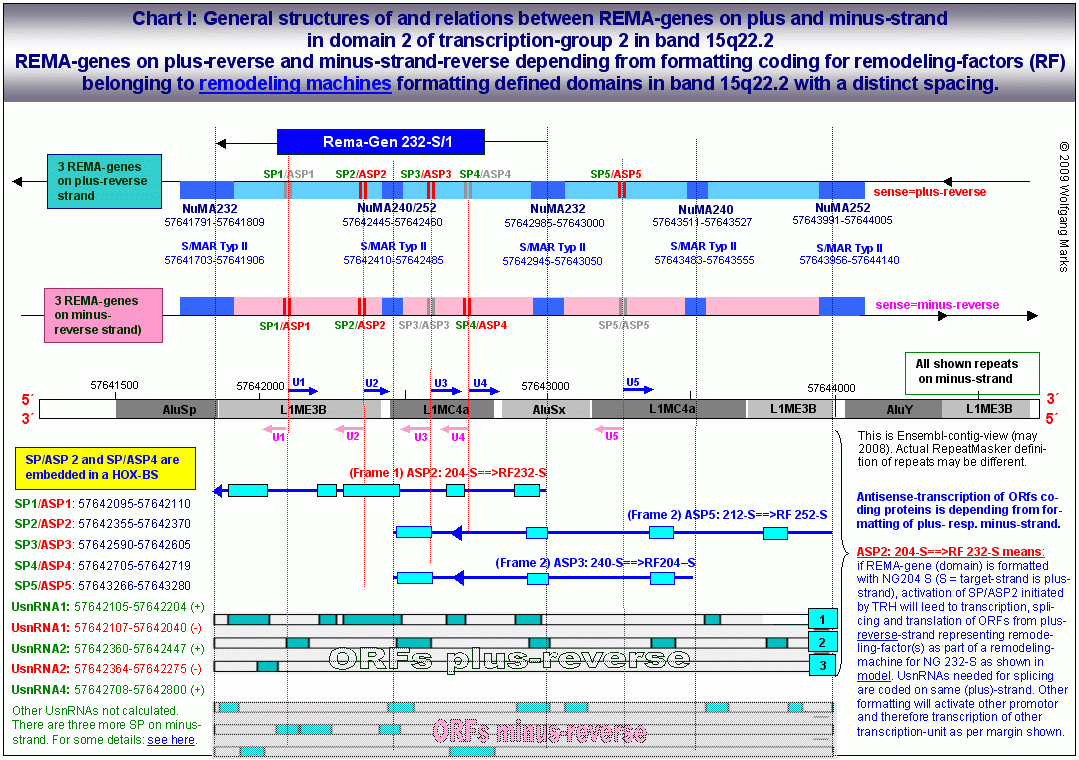
Die Abbildungsreihe: General structures.. zeigt eine computational analysis mehrerer DNA-Abschnitte mit Clustern von REMA-Genen auf dem plus-reverse und dem minus-reverse-Strang der oben angesprochenen Domäne 2, die jeweils von SMARs der Klasse II79 begrenzt werden. Jeder dieser REMA-Gen-Cluster enthält mehrere REMA-Gene die detaillierte Struktur eines solchen Clusters 1 zeigt das Chart I.
|
|
|
|
|
|
|
|
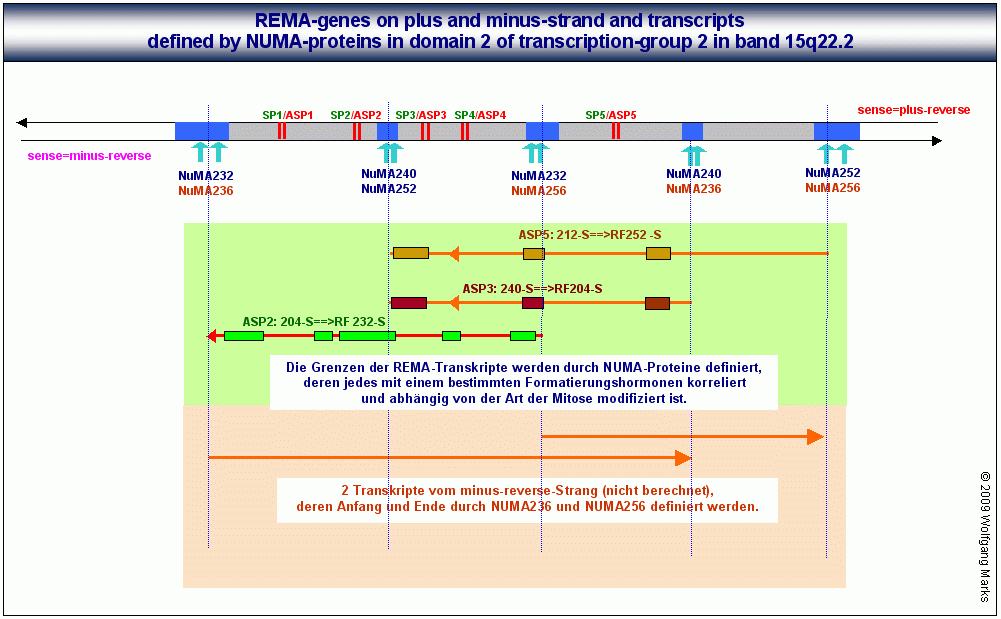
Wie in diesem Chart I zu sehen ist, bilden die sense- Promotoren von UsnRNAs und die antisense-Promotoren von REMA-Genen Paare. Zumindest in den von mir bis dato analysierten REMA-Genen ist jeder antisense-Promotor zugleich ein sense-Promotor (das ist aber keine Regel, die für das ganze Genom gilt) nicht jeder sense-Promotor aber auch zugleich ein antisense-Promotor, da es nach meinen Analysen mehr sense- als antisense-Promotoren gibt. Da das Transkript, das durch einen antisense-Promotor aktiviert wird, in der Regel mehrere Exons/ORFs enthält, für deren Spleissen eine entsprechende Anzahl von UsnRNAs benötigt wird, ist leicht einzusehen, dass die Zahl der sense-Promotoren grösser sein muss, als die Zahl der antisense-Promotoren. Antisense-Promotoren von REMA-Genen sind anscheinend darüberhinaus fähig, sowohl eine antisense-Transkription vom plus- als auch vom minus-Strang zu initiieren ob ein bestimmter ASP die antisense-Transkription vom plus- oder vom minus-Strang einleitet, hängt wahrscheinlich allein vom Vorhandensein und von der Zusammensetzung eines spezifischen Aktivierungs- bzw. Transkriptionskomplexes ab.
Der REMA-Gen-Cluster auf Chart I enthält insgesamt 3 REMA-Gene auf dem plus-reverse- und 2 REMA-Gene auf dem minus-reverse-Strang. Die auf dem plus-reverse- und dem minus-reverse-Strang kodierten ORFs repräsentieren demnach fünf unterschiedliche Transkriptionseinheiten, die durch jeweils eine S/MAR genauer: durch eine Bindestelle für ein spezifisches NUMA-Protein begrenzt werden. Jedes der fünf Transkripte von denen ich nur die drei des plus-reverse-Strangs zeige - ist mit einem der fünf antisense-Promotoren verknüpft. Es fällt auf, dass diese Transkriptionseinheiten wie Exon/Intron-Gene von mir polycistronische Gene genannt organisiert sind: sie werden höchstwahrscheinlich auch wie diese transkribiert und translatiert mit dem Unterschied, dass der Promotor des jeweiligen REMA-Gens innerhalb desselben liegt und die Grenzen des REMA-Gens nicht durch Promotoren und poly-A-Signale, sondern durch S/MARs und NUMA-Bindestellen bestimmt werden.
|
|
|
|
Abb.:In jeder SMAR innerhalb eines REMA- Gen-Clusters gibt es mehrere Bindestellen für NUMA-Proteine. NUMA-Proteine lassen sich Formatierungshormonen und Nukleosomengrössen zuordnen.
Also gibt es ebensoviele verschiedene NUMA-Proteine, wie es transkriptionsaktive Formatierungshormone gibt.
|
|
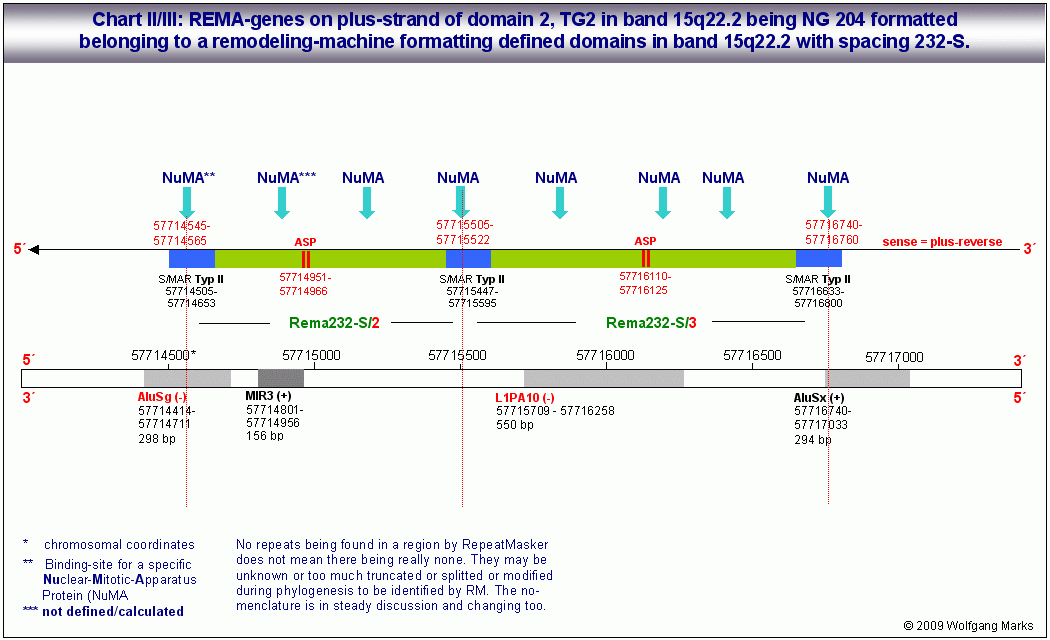
Die Expression des REMA-Gens 232-S/1 ist allein abhängig von der Formatierung der Domäne mit der Nukleosomengrösse 232 denn nur bei dieser Formatierung liegt der Promotor SP2/ASP2 auf der linker-DNA zwischen zwei Nukleosomen-Cores. Das Gen selbst besteht aus fünf ORFs bzw. Exons, die nach der Transkription wahrscheinlich zu zwei verschiedenen messenger-RNAs gespleisst werden, die jeweils einen remodeling-Faktor kodieren.
Dies geschieht mit Hilfe von ubiquitären UsnRNAs bzw. UsnRNPs (auch SNURPS genannt), deren Gene (zumindest für die major snRNAs U1, U2, U3, U4/U6, U5) in unterschiedlich grosser Zahl über das Genom verstreut sind. Allein für die U1-snRNA muss es nach meinen Berechnungen ca. 130 verschiedene Gene geben, also solche, die sich in ihrer Länge und/oder ihrer Sequenz unterscheiden. U1-snRNA ist also nicht gleich U1-snRNA - das sollte auch für die anderen Mitglieder der UsnRNA-Familie gelten. Zusätzliche Variabilität erhalten UsnRNAs durch den Zusammenschluss mit Proteinen zu sogenannten SNURPS. Es kann deshalb auch nicht gleichgültig sein, welche UsnRNAs bei der Transkription eines bestimmten Gens aktiviert werden gleichgültig, ob es sich dabei um ein Gen handelt, das eine RNA oder eines, das eine mRNA kodiert.
Darüberhinaus ist die Verteilung der UsnRNA-Gene nach meinen Berechnungen auch nicht zufällig, sondern mit der Anordnung solcher Gene korreliert, die mit ihrer Hilfe gespleisst werden. Dies trifft wie ich in Teil III dieser Arbeit zeigen werde - auf polycistronische metabole Gene ebenso zu wie auf ALU-Gene und die hier zur Diskussion stehenden REMA-Gene.
Bevor REMA-Gene transkribiert werden können, muss das Histon H1, das die Transkription der Gene verhindern würde, zunächst durch ein Surrogat ersetzt werden. Dieses Surrogat bildet der von mir H1PAC80 genannte Komplex aus einem formatierungshormonabhängigen HMGN-Protein, das einen HRPK81, ein Molekül Ubiquitin und ein Molekül Nukleoplasmin gebunden hat. Die Bildung dieses Komplexes und seine Funktion wird in Teil III im Kapitel Der Weg zur Transkriptionsschleife im Detail erläutert. Ich gehe deshalb an dieser Stelle nicht weiter darauf ein.
Auch die Transkription der UsnRNA-und REMA-Gene und der Mechanismus der Promotor-Aktivierung soll hier nur in groben Zügen diskutiert werden, da diese Prozesse in weiten Teilen mit den Abläufen übereinstimmen, wie sie in Teil III für metabole Gene ausführlich und Schritt für Schritt besprochen werden.
Die Transkription von UsnRNA und REMA-Genen.
Die Zusammensetzung der humanen UsnRNA-Promotoren ist im Grossen und Ganzen, von einigen Ungewissheiten abgesehen, bekannt. Nouria Hernandez schreibt:82
The snRNA promoters in vertebrates, A. thaliana, D. melanogaster, and sea urchins all contain an element, variously called the PSE, PSEA, or USE, centered 50 to 70 bp upstream of the transcription start site. The factor binding to this element has been best characterized in the human system, and is variously known as PBP, PTF, or SNAPc. It is a complex containing five types of subunits, SNAP190, SNAP50 (PTFb), SNAP45 (PTFd), SNAP43 (PTFg), and SNAP19 (see (8) and references within). SNAP190 forms the backbone of the complex, with SNAP19 and SNAP45 associating toward the N and Cterminus,respectively, of the molecule. SNAP43 can associate with the same region of SNAP190 as SNAP19, and SNAP50 joins the complex by associating with SNAP43 (see Figure 3 below for an illustration SNAPc).[...]
In the case of the pol II snRNA promoters, the transcription initiation complex has not yet been assembled in a stepwise fashion in vitro, but many of its components have been identified functionally by depletion of transcription extracts with specific antibodies and reconstitution of transcription with recombinant factors. Thus, many of the players are known but their mode of assembly on snRNA promoters is not, and thus their location in Figure 2A is arbitrary. Depletion and reconstitution experiments indicate that recombinant TBP (but not the TBPcontaining complexes TFIID or TFIIIB), TFIIB, TFIIA, TFIIF, and TFIIE are required ((14), and references therein). TFIIA does not require TFIIH, or requires much lower levels than transcription from a mRNA promo- ter. If TFIIH is indeed not required, this raises the interesting question of how open complex formation is achieved at snRNA promoters. A combination of all the general transcription factors and SNAPc does not initiate transcription from the U1 promoter,suggesting that additional as yet unidentified factors are required (14).
Die Initiation der Transkription eines UsnRNA-Gens bedarf also im Prinzip nur eines einzigen Elements, eben des PSE. Die Zusammensetzung des Transkriptionskomplexes selbst ist unklar, im Prinzip sind zwar die gleichen TFII-Faktoren beteiligt, wie bei der Transkription der mRNA-kodierenden Gene, aber ihre spezifische Funktion und Assemblierung konnte bis heute nicht vollständig aufgeklärt werden. Und schliesslich werden ausser den genannten TFs offensichtlich weitere Faktoren benötigt, um die Transkription einer UsnRNA einzuleiten. Zu diesen Faktoren gehören wahrscheinlich bestimmte Architekturproteine wie solche der ARBP/MeCP2-, SAFA/B- oder der SATB-Familie, ein NuMA-Protein und ein spezifischer Hormon-Rezeptorproteinkomplex, wie er auch für die Transkription metaboler Gene erforderlich ist. Dies schliesse ich unter anderem daraus, dass SMARs, die REMA-Gene und ALU-Gene begrenzen (SMARs der Klasse II)83, Bindestellen für diese Proteine aufweisen.
Die Transkription eines UsnRNA-Gens wird wahrscheinlich eingeleitet durch Bindung der stromab gelegenen SMAR II an die Kernmatrix unter Vermittlung von Proteinen der ARBP/MeCP2-, der SAF- und vor allem der NuMA-Gruppe84. Dadurch wird der Promotor, an den zwischenzeitlich ein spezifischer HRPK und ein SATB-Protein gebunden hat, in die unmittelbare Nähe der Matrix gebracht und mit Hilfe der GTFs an der SMAR II/Matrix fixiert. Die Bindung der RNAP II vervollständigt den Transkriptionskomplex.
Promotor und SMAR bilden nun eine Transkriptionsschleife, in der das UsnRNA-Gen beliebig oft transkribiert werden kann.85 Dieser Ablauf wiederholt sich für jedes einzelne UsnRNA-Gen, das über seinen Promotor aktiviert wurde.
Die Transkription eines REMA-Gens beginnt mit der Bindung spezifischer Architekturproteine an zwei SMARs, die Bindestellen für diese Proteine enthalten. Dies sind naturgemäss die gleichen Proteine, die auch bei der Transkription der UsnRNA-Gene die Funktion haben, die SMAR II an die Kernmatrix zu binden, also Proteine der SATB-, NuMA- und SAF-Gruppe. Die stromab gelegene SMAR II bindet wahrscheinlich zuerst an die Matrix, dann bindet die stromauf gelegene SMAR II an die gleiche Stelle die beiden SMARs der gleichen Klasse bilden also einen Komplex. Dieser Prozess könnte durch das sogenannte mass-binding katalysiert werden, das für SAF-Proteine beschrieben wurde. Darauf komme ich bei der Besprechung der Architekturproteine in Teil III noch einmal zurück.
|
|
|
|
|
Durch Bindung des Antisense-Promotors an die Matrix und die beiden SMARs bildet sich eine butterfly-Struktur. Nachdem die RNAPII an den PIC rekutiert wurde, löst sich der Promotor wieder von der Matrix und die Transkriptionsschleife - die jetzt von SMAR zu SMAR reicht und alle ORFs/Exons des REMA-Gens umfasst - wird transkribiert.
|
|
|
Anschliessend wird der antisense-Promotor (der durch die Formatierung der Domäne mit einer bestimmten NG determiniert ist) mit Hilfe eines zellspezifischen Hormon-Rezeptorproteinkomplexes und eines Proteins der ARBP-Familie an die beiden SMARs und damit auch an die Matrix gebunden. Dadurch entsteht zunächst die in der Skizze gezeigte Struktur, die einem Schmetterlingsflügel ähnlich sieht. Nach Bindung der GTFs und der RNAPII und Einleitung der Transkription löst sich der Promotor wieder von der Matrix und es formiert sich die Transkriptionsschleife, die von SMAR II zu SMAR II reicht und jetzt das REMA-Gen mit seinen Exons/ORFs umfasst.
Das REMA-Gen wird transkribiert, die Exons wie bei einem normalen metabolischen Gen mit Hilfe der zuvor generierten UsnRNAs cotranskriptionell gespleisst, die gespleisste messenger-RNA ins Zytoplasma transportiert, dort translatiert und gegebenenfalls weiterverarbeitet. Dies können spezifische Modifikationen sein, aber auch eine Aufspaltung des Polypeptids in zwei verschiedene remodeling-Faktoren, die sich mit anderen zeitgleich im Chromosomenband generierten remodeling-Faktoren zu einer für die Zelle und den jeweiligen Entwicklungsstand spezifischen remodeling-Maschine zusammenschliessen.
Der oben beschriebene Vorgang läuft zeitgleich im gesamten Chromosomenband in allen Chromatin-Domänen ab, die durch die Nukleosomengrösse 232-S zu einer Transkriptionseinheit, zu einem Netzwerk zusammengefasst sind.
Remodeling-Maschinen für die Markierungsgrössen 204, 208, 212 werden während des prä-mitotischen Zellzyklus, solche für transkriptionsaktive Nukleosomengrössen und die Grösse 150 in der post-mitotischen Phase nach der Zellteilung aktiviert.
Wie ich zuvor bereits habe anklingen lassen, bin ich der festen Überzeugung, dass die Differenzierung einer Zelle nicht in einem Schritt ablaufen kann, sondern nach der Mitose einen zweiten Zellzyklus erfordert, der allerdings nicht mit einer erneuten Zellteilung, sondern in die Arbeitsphase/Interphase mündet.86 Obwohl ich auf Details dieses Prozesses erst am Ende dieses Teils II bei der Vorstellung meines 2-Zellzyklen-Modells der Zelldifferenzierung eingehe, will ich an dieser Stelle aber doch kurz erläutern, wie die Beschriftung der drei gezeigten Transkriptionseinheiten aus Chart I zu lesen ist.
In der G0 und auch in der Interphase sind die Chromatin-Domänen einer definierten Zelle entweder inaktiviert das würde in meinem Modell bedeuten, dass sie mit der NG 150 formatiert und die Nukleosomen mit H1 verschlossen und gegebenenfalls zu einer Chromatinstruktur höherer Ordnung kondensiert sind oder sie sind mit einer der 19 transkriptionsaktiven Nukleosomengrössen formatiert.87 Da die Aktivierung des ASP eines REMA-Gens mit der Formatierung der jeweiligen Domäne mit einer bestimmten Nukleosomengrösse verknüpft ist, habe ich im gezeigten Ausschnitt aus dem Chart I bei jedem Transkript zunächst die Nukleosomengrösse angegeben, durch die der zugehörige ASP aktiviert werden kann (die Aktivierungsgröße) und dann die Zielgrösse angegeben, also die Nukleosomengrösse, welche die aus den remodeling-Faktoren gebildete remodeling-Maschine programmieren kann.
|
|
|
|
|
ASP2: 204-S==>RF 232-S bedeutet demnach: Der ASP2 ist aktivierbar, wenn die Domäne mit der NG 204 formatiert ist (er liegt dann auf der linker-DNA zwischen zwei Nukleosomenkernen). Der ASP 2 aktiviert die Transkription einer RNA aus 5 ORFs. Nach dem Spleissen und der Translation repräsentieren die Proteine remodeling-Faktoren für die NG 232-S.
Für die gezeigten 3 Transkripte ergeben sich damit folgende Interpretationsmöglichkeiten:
Fall 1a (ASP5):
Die Zelle differenziert sich:
Die betreffende Domäne der Mutterzelle wird in der G2-Phase des prämitotischen Zellzyklus mit der NG 212-S formatiert (Markierung für Format kopieren) und in den Tochterzellen nach der Mitose wieder mit der Ausgangs-NG der Mutterzelle formatiert. In Nukleosomengrössen geschrieben:
M2: 252-S -> 212-S
M1: 212-S -> 252-S
Der ASP5 in Chart I muss ein Promotor sein, der in der postmitotischen Phase in den Tochterzellen durch die Formatierung der Domäne(n) mit der NG 212-S (Format kopieren) aktiviert wird. Das Transkript des REMA-Gens das aus vier ORFs besteht kodiert remodeling-Faktoren für die NG252-S. Nach Durchlaufen des postmitotischen Zellzyklus sind die Formate dieser Domäne in Mutter- und Tochterzellen damit wieder identisch.
Fall 1b: (nicht gezeigt)
Die Zelle proliferiert das Format aller Domänen wird kopiert:
Die Nukleosomenkonfiguration der Domäne wird während der Replikation der DNA in der S-Phase der Mitose zunächst kopiert und in der G2-Phase zellspezifisch modifiziert:
M1:252-S -> 252-S
Fall 2 a und b:
Das Format einer Domäne soll geändert werden:
Die Domäne der Mutterzelle wird prä-mitotisch mit der NG 204 formatiert (markiert) und in der post-mitotischen Phase in den Tochterzellen mit einer neuen NG formatiert. In Nukleosomengrössen geschrieben:
M1:alte NG ->204-S
M2: 204-S->neue NG
Fall 2a (ASP3):
Markierung für Formatierung ändern während des prämitotischen Zellzyklus:
Der ASP3 in Chart I muss ein Promotor sein, der in der Interphase mit einer transkriptionsaktiven Nukleosomengrösse (zum Beispiel 240) formatiert war. Das Zellprogramm sieht für diese Domäne eine Änderung der Nukleosomengrösse vor. Das Transkript des REMA-Gens das aus drei ORFs besteht kodiert dementsprechend remodeling-Faktoren für die NG 204-S. Mit dieser NG wird die Domäne während des prämitotischen Zellzyklus formatiert. Nach der Zellteilung führt die Formatierung der Domäne mit der NG 204-S zur Aktivierung von spezifischen ASP und zur Synthese von remodeling-Faktoren für eine neue epigenetisch destinierte NG.
Fall 2b (ASP2):
Markierung für Formatierung ändern (im postmitotischen Zellzyklus):
Der ASP2 wird durch Formatierung der Domäne mit der NG 204 im postmitotischen Zellzyklus aktiviert. Seine Aktivierung führt zur Transkription und Translation von remodeling-Faktoren für die NG 232-S. Nach Beendigung des postmitotischen Zellzyklus ist die Domäne also mit der NG 232-S formatiert.
Jeder REMA-Gen-Promotor ist also Teil eines epigenetischen Formatierungssystems, in der die Formatierung einer Domäne die zukünftige Formatierung dieser gleichen Domäne bereits beinhaltet. Oder anders ausgedrückt: jedes REMA-Gen hat eine bestimmte Vergangenheit, eine bekannte Gegenwart und eine definierte Zukunft. Oder in der Sprache der Programmierer:
ASP von REMA-Genen sind Zeiger, auch Sprungadressen genannt, die auf den jeweils nächsten Programmteil, hier die nächste epigenetisch definierte Nukleosomengrösse verweisen.
Die Vorgänge während der prä- und postmitotischen Zellzyklen werde ich am Schluss dieses Teils II noch im Detail besprechen.
|
|
75Über die Einteilung der DNA in Transkriptionsgruppen, Chromatin-Domänen und subdomänen und die Definition dieser Bereiche wird weiter unten ausführlich gesprochen.
76SMARs der Klsse II unterteilen REMA- und ALU-Gene in Transkriptioneinheiten.
77Für das Spleissen der DNA Transkripte von polycistronischen Genen (Exon/Intron-Gene) werden kleine RNAs benötigt, die man nach ihrer Funktion beim Spleißprozess in sechs verschiedene Klassen einteilt: U1-, U2- (U3-), U4-, U5- und U6snRNA. Jede dieser kleinen RNAs hat eine definierte Funktion im Spleissosom. Auch REMA-antisense-Transkripte werden mit Hilfe solcher snRNAs gespleisst.
78Das hier angesprochene Chromatin-remodeling findet während der Mitose statt. Das werde ich in dieser Arbeit noch ausführlich referieren.
79Die Einteilung der SMARs in bestimmte Klassen werde ich später noch erläutern.
80H1 Push Aside Complex - darüber wird im Kapitel: Vom Chromomer zum Transkriptosom noch ausführlich gesprochen.
81Hormon-Rezeptorproteinkomplex, Komplex aus einem Rezeptorprotein und einem Hormon, der nach der Bindung/Aktivierung in Untereinheiten zerfällt.
82snRNA genes: a model system to study fundamental mechanisms of transcription;
Nouria Hernandez; The Journal of Biological Chemistry; JBC Papers in Press. Published on June 4, 2001; http://www.jbc.org/cgi/reprint/R100032200v1.pdf
83Über die Struktur und die Klassifizierung von SMARs werde ich im Anschluss an dieses Kapitel sprechen.
84SATB- und HMGA-Proteine, die bei der Initiation der mRNA-Gene eine wichtige Rolle spielen, sind an diesem Prozess mit einiger Sicherheit ebensowenig beteiligt, wie an der Transkription der REMA-Gene.
85Vor der Initiation der Transkription wurden die linker-Histone, die zu diesem Zeitpunkt aus einer H1-Variante bestehen, gegen einen HMGN-Hormon-Rezeptorproteinkomplex ausgetauscht siehe: Der postmitotische Zellzyklus und Teil III dieser Arbeit.
86Auch die proliferative Teilung einer nicht ausdifferenzierten Zelle während der Ontogenese erfordert in meinem Modell der formatierten DNA zwei Zellzyklen. Lediglich die proliferative Teilung einer ausdifferenzierten Zelle lässt sich danach in einer Mitose und in einem Zellzyklus abwickeln.
87Von dieser Regel sind bestimmte Regionen, so zum Beispiel die Zentromere undTelomere ausgenommen.
|
|
|