|
Stillegung und Imprinting - zwei Seiten der gleichen Medaille.
Auch die Stillegung einer Chromatin-Domäne ist ein epigenetisch definierter Prozess, der den gleichen Regeln folgt, wie die Programmierung einer Chromatin-Domäne mit einer neuen aktiven Nukleosomengrösse. Die Formatierung mit der Nukleosomengrösse 150 erfolgt, wie in meinem 2-Zellzyklen-Modell beschrieben, in einem ersten Schritt durch Markierung der stillzulegenden Domäne mit der Nukleosomengrösse 208 im prämitotischen Zellzyklus. Dabei entsprechen die Vorgänge im Prinzip denen bei der Formatierung einer Domäne mit der NG 204 oder 212. Erst während des postmitotischen Zellzyklus wird die Domäne mit der Nukleosomengrösse 150 formatiert und die Oktamere mit modifziertem H1 unlöslich an die DNA gebunden.
Es liegt auf der Hand, dass die Aktivierung von remodeling-Maschinen sowohl für die Markierung der Domäne mit der NG 208 im prämitotischen Zellzyklus, als auch die Formatierung der Domäne mit der NG 150, die sich an die Zellteilung anschliesst und zur Inaktivierung der Domäne und zur Bindung der Nukleosomen-Cores an ein modifiziertes Histon H1 führt, nicht von einem Formatierungshormon abhängig oder gesteuert sein kann. Eine Möglichkeit wäre, dass die jeweiligen Komplexe anstatt eines transkriptionsaktiven Hormons ein spezifisches Stillegungshormon oder einen Silencingfactor enthalten, der die selektive Bindung an REMA-Gen-Promotoren für die NG 208 und 150 gewährleistet. Ein zweite Möglichkeit wäre, dass es gerade die Abwesenheit der hormonellen Faktoren oder spezifischer HRPKs ist, welche die Spezifität der Transkriptionskomplexe für REMA-Gen-Promotoren, die mit der NG 208 und 150 verknüpft sind, zur Folge hat.
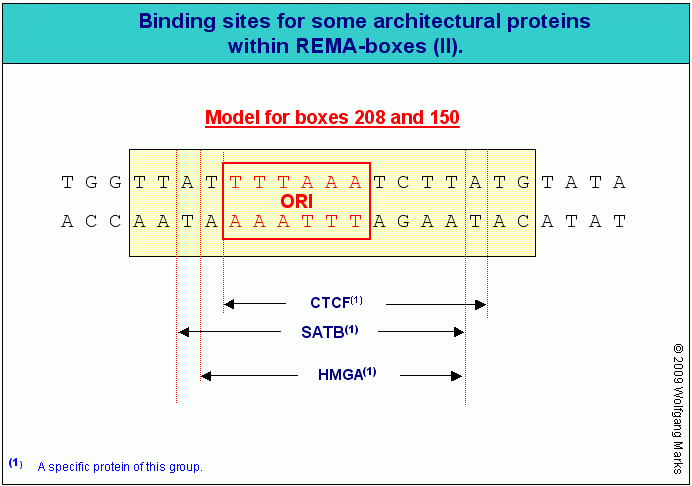
Wie ich im Kapitel REMAKES ausgeführt habe, wird eine aktive Chromatin-Domäne von einer bestimmten Startstelle aus formatiert, die in einer engen topografischen und funktionellen Beziehung zu einer definierten REMA-Box innerhalb einer SMAR III, eines REMAKEs steht. Die Bindung der remodeling-Maschine an die REMA-Box und die Definition der Startstelle wird durch einen Komplex vermittelt, der abhängig von der Entwicklungsstufe, auf der sich die Zelle befindet, Proteine der SAF-, der ARBP-, SATB-, HMGN-, HMGA oder HMGB-Familie sowie als massgebliches Agens einen Komplex aus einem Rezeptorprotein und einem primären Transkriptionshormon enthält. Würde man einen solchen Formatierungskomplex nicht postulieren, müsste die spezifische Formatierungs-Startstelle einzig und allein durch eine bestimmte remodeling-Maschine definiert sein es müsste dann aber statt einer einzigen definierten REMA-Box für eine formatierungshormonspezifische remodeling-Maschine zum Beispiel der NG 232 eine ganze Reihe von docking stations für solche remodeling-Maschinen der NG 232 geben. Das ist aber da die REMA-Box auch Bindestellen für verschiedene Architekturproteine enthält und zugleich den Origin Of Replication (ORI) darstellt, nur schlecht vorstellbar und stimmt auch nicht mit dem Ergebnis meiner Computer-Analysen von REMAKEs überein, die ich in Teil II präsentiert habe.
Die logische Folgerung aus dem Vorgetragenen ist, dass es auch für die Stillegung von Chromatin-Domänen einen solchen Aktivierungskomplex geben muss, der die Formatierungs-Startstelle für die Formatierung mit der Nukleosomengrösse 208 bzw. 150 unter Mitwirkung eines Silencingfaktors festlegt.
Die Inaktivierung von Genen und Gen-Allelen mit Hilfe von
silencing- und imprinting-Faktoren.
Die Frage, auf welchem Wege die Zelle einzelne Transkripte, Gene, Chromatin-Domänen oder ganze Transkriptionsgrupppen stillegt, ist vor allem deshalb von ausserordentlichem Interesse, weil Fehler im Ablauf dieser Prozesse zwangsläufig zu schwerwiegenden Störungen im Metabolismus der Zelle und des ganzen Organismus führen.
Nach dem augenblicklichen Stand des Wissens nutzt die Zelle (zumindest) fünf verschiedene Mechanismen, um Gene oder Gengruppen und Transkripte oder Transkriptgruppen abzuschalten oder ihre Expression zu modulieren.
1. Inaktivierung eines Gens oder einer Gengruppe durch Formatierung der betreffenden Chromatin-Domäne mit der NG150. Dieser Prozess umfasst beide Allele.
2. Imprinting - Inaktivierung eines mütterlichen oder väterlichen Gen-Allels.
3. Steuerung der Genexpression durch das Hox-Gensystem.
4. RNA-Interferenz - die selektive Eliminierung einer messenger-RNA nach der Transkription.
5. RNA-editing posttranskriptionelle Veränderung einer messenger-RNA durch chemische Modifikation einzelner
Nukleotide der RNA.
An dieser Stelle werde ich mich ausschliesslich mit der Inaktivierung von Genen und dem Imprinting beschäftigen, das nach dem Ergebnis meiner Analysen eng mit dem Prozess der Inaktivierung verknüpft ist. Zwar ist die RNA-Interferenz - die Eliminierung spezifischer messenger-RNAs durch kleine RNAs, die man miRNA (microRNA), siRNA oder auch piRNA nennt - ein in jüngerer Zeit vielbeachteter Mechanismus zur Modulation der Genexpression - da dieses Ausschalten von Gen-Transkripten mit Hilfe der RNA-Basenpaarung aber auf der posttranskriptionellen Ebene geschieht, werde ich mich in dieser Arbeit nicht weiter mit diesem Mechanismus beschäftigen.
Gleiches gilt für das RNA-editing, das zwischen Transkription und Translation stattfindet und deshalb hier auch nicht weiter untersucht werden soll.
Die Steuerung der Genexpression durch Hox-Gene werde ich am Ende des Teils III diskutieren, da diese Steuerung unter anderem auf der globalen, genomweiten Regulation zweier Gensysteme beruht, von denen ich das eine das REMA-Gen-System, zwar schon vorgestellt habe, das andere aber das ALU-Gen-System - erst in Teil III meiner Arbeit besprechen werde.
Schließlich gibt es für einzelne Gene152 und Gengruppen Regelkreise, welche die Genfunktion abhängig von zellinternen oder -externen Einflüssen oder vom Vorhandensein bestimmter Substrate zu steuern in der Lage sind.
Ich werde mich also jetzt mit der Frage beschäftigen, welche Faktoren an der Inaktivierung und dem Imprinting eines Gens beteiligt und wie die Prozesse beschaffen sein könnten, die diese Vorgänge steuern. Um einer einheitlichen Sprachregelung willen153 und um Missverständnisse zu vermeiden, definiere ich Stillegung (Synonyme: Inaktivierung, Silencing) von nun an als Inaktivierung beider Allele eines Gens Imprinting demgegenüber als die Inaktivierung eines einzelnen mütterlichen oder väterlichen Genallels.
Gen-Stillegung unter Mitwirkung von Proteinen der HMGA-
und der SATB-Familie.
Bis etwa zum 80. Tag sind nach dem Ergebnis meiner Analysen Proteine der HMGA-Familie für die Stillegung von Genen zuständig. Nach diesem Zeitpunkt sind es Proteine der SATB-Gruppe, von denen zwei seit geraumer Zeit bekannt sind.
HMGA-Proteine als Silencing-Faktoren
Die HMGA-Familie besteht nach meinen Berechnungen aus insgesamt 19 Proteinen, von denen jedes mit einem Formatierungshormon und damit auch mit einer der 19 transkriptionsaktiven Nukleosomengrössen korreliert ist.
HMGA-Proteine sollen bevorzugt an die kleine Furche der B-DNA binden und dies vor allem in AT-reichen155 Abschnitten. Die Bindung an die DNA soll von drei AT-Hook-DNA-Bindungsmotiven vermittelt werden, wie diese Bindung etabliert wird, ist aber nicht mit Bestimmtheit gewußt. Entscheidender als die Bindungssequenz der Ziel-DNA soll die dreidimensionale Struktur der DNA sein: anscheinend binden HMGA-Proteine bevorzugt an verdrehte oder geknickte DNA.
Durch Bindung von HMGA-Proteinen kann lineare DNA verbogen werden vice versa können Verbiegungen in verbogener oder geknickter DNA durch HMGA-Proteine auch wieder entfernt werden. Ausserdem kann durch Bindung von HMGA-Proteinen DNA partiell entwunden oder zu Schleifen geformt werden. Dieses Phänomen, das uns später noch interessieren wird, wurde m.W. zuerst von Lehn et al. (1988), später auch von Falvo et al. ( 1995),Chase et al. (1999), Bagga et al. und Liu et al. (jeweils 2000) beschrieben. Für die Konformationsänderung der DNA wurden unterschiedliche Mechanismen als mögliche Ursachen angeführt: in der Diskussion ist sowohl das Andocken der drei AT-Hooks eines einzelnen HMGA-Moleküls an weit voneinander entfernte Bereiche der Ziel-DNA, als auch die koordinierte Bindung mehrerer Moleküle an bestimmte Bereiche der DNA und ein darauf folgendes nicht kovalentes cross-linking (Reeves, 2001).
HMGA-Proteine werden nach ihrer Translation im Zytoplasma im Zellkern vielfach chemisch modifiziert: Phosphorylierung, Azetylierung, Methylierung und Poly-ADP-Ribosylierung ist beschrieben worden (Zusammenfassung 2001: Reeves und Beckerbauer). Phosphorylierung und Azetylierung scheinen in vitro die Bindungseigenschaften und die biologische Aktivität von HMGA-Proteinen zu beeinflussen (Nissen et al. (1991); Reeves et al. (1991). Die Phosphorylierung von HMGA-Proteinen scheint abhängig vom Zellzyklus zu sein und wird sowohl von endogenen zellulären Signalen kontrolliert (Detivaud et al. (1999), als auch von Signalereignissen an oder in der Zellmembran beeinflusst (Wang et al., 1995; Banks et al., 2000; Xiao et al., 2000.
Disney et al. (1989) und Reeves (1992) sowie Saitoh und Laemmli (1994) haben beschrieben, dass HMGA-Proteine aktiv an der dynamischen Änderung der Chromatinstruktur während der Chromosomenkondensation zum Zwecke der Zellteilung beteiligt sind. HMGA-Proteine haben aber auch einen direkten Einfluss auf die Genexpression: die durch HMGA induzierte Elimination von DNA-Verbiegungen steht in direktem Zusammenhang mit der Rekrutierung von Transkriptionsfaktoren (Thanos und Maniatis, 1995) und remodeling-Maschinen, so zum Beispiel des SWI/SNF-Komplexes (Agresti und Bianchi, 2003).
HMGA wird in der frühen Phase der Ontogenese exprimiert.
Die bis jetzt bekannten HMGA-Proteine werden überwiegend in nicht ausdifferenzierten Zellen oder Embryonalzellen exprimiert. In ausdifferenzierten Zellen ist nur wenig HMGA nachweisbar (Bustin und Reeves, 1996); Chiapetta et al. 1996). Eine Überexpression von HMGA-Proteinen ist ebenso mit malignen Prozessen verbunden wie das Fehlen des Proteins. Bei einer HMGA2-knock-out-Maus führt das Fehlen des Proteins zu Zwergenwachstum bei der Überexpression von HMGA2 werden vermehrt Fettzellen gebildet, was nach Meinung von Anand und Chada (2000) darauf hinweisen könnte, dass HMGA2 eine Rolle bei der Adipogenese spielt.
Schließlich sind HMGA-Proteine in vielen malignen Tumoren überexprimiert (u.a. bei Brust-, Prostata-, Colon-, Magen-, Haut und Lungenkrebs (Reeves und Beckerbauer, 2001), aber auch in benignen Bindegewebstumoren ist eine abnorme Produktion von HMGA(2) Proteinen zu beobachten (Battista et al., 1999; Arlotta et al., 2000).
SATB-Proteine als Silencing-Faktoren.
SATB1und SATB2 gehören nach dem Ergebnis meiner Untersuchungen mittels IMPACD zu einer Proteinfamilie, die insgesamt 30 Proteine umfasst und durch 4 verschiedene Gene kodiert wird, die sich nach meinen Analysen einschliesslich zahlreicher Kopien auf den Chromosomen 2, 3, 5 und 13, 14, 15, 18,19 und 20 befinden.
Das von der Gruppe um Terumi Kohwi-Shigematsu entdeckte Protein SATB1 scheint ein Molekül zu sein, das nur in bestimmten Zellinien vorkommt, so in Thymozyten, aktivierten T-Zellen und in einigen Vorläufern der genannten Zellen. Wenn die Expression dieses Proteins im Maus-Modell verhindert wird, wird die Regulation vieler Gene in Thymozyten und damit die Entwicklung von T-Zellen gestört. SATB1 soll weiter eine wichtige Rolle bei der Organisation der DNA in loops spielen, das heisst in chromosomale Schleifen, die in unregelmäßigen Abständen an die Kernmatrix angeheftet sind: SATB1 soll dabei eine spezifische DNA-Sequenz erkennen, genannt BUR (Base Unpairing Region) die als charakteristisch für DNA-Bereiche gilt, in denen die DNA mit der Kernmatrix in Kontakt steht, also für die weiter oben (in Teil II) charakterisierten SMARs. SATB1 soll aktiv an der Bildung dieser Chromatin-Schleifen oder schlaufen (loops) beteiligt sein, indem es BUR-DNA-Sequenzen wie eine biologische Klammer miteinander verbindet. Darüber hinaus soll SATB1 auch mit der Rekrutierung von remodeling-Faktoren oder -komplexen wie dem NURD-, dem CHRAC und auch dem ACF-Komplex korreliert sein, was eine Erklärung für die Beobachtung bieten soll, dass SATB1 offensichtlich Einfluss auf die Expression von Genen hat.156 Terumi Kohwi-Shigematsu et. al. schreiben im abstract zu ihrer Arbeit:
Eukaryotic chromosomes are organized inside the nucleus in such a way that only a subset of the genome is expressed in any given cell type, but the details of this organization are largely unknown. SATB1 ('special AT-rich sequence binding 1'), a protein found predominantly in thymocytes, regulates genes by folding chromatin into loop domains, tethering specialized DNA elements to an SATB1 network structure. Ablation of SATB1 by gene targeting results in temporal and spatial mis-expression of numerous genes and arrested T-cell development, suggesting that SATB1 is a cell-type specific global gene regulator. Here we show that SATB1 targets chromatin remodeling to the IL-2R ('interleukin-2 receptor ') gene, which is ectopically transcribed in SATB1 null thymocytes. SATB1 recruits the histone deacetylase contained in the NURD chromatin remodeling complex to a SATB1-bound site in the IL-2R locus, and mediates the specific deacetylation of histones in a large domain within the locus. SATB1 also targets ACF1 and ISWI, subunits of CHRAC and ACF nucleosome mobilizing complexes, to this specific site and regulates nucleosome positioning over seven kilobases. SATB1 defines a class of transcriptional regulators that function as a 'landing platform' for several chromatin remodeling enzymes and hence regulate large chromatin domains.
In SATB1 -/- Thymocyten verändert sich das Assoziationsverhalten SATB1-bindender DNA Elemente: die normalerweise an der Kernmatrix lokalisierten SATB1-bindenden DNA-Fragmente verlieren ihren Kontakt zur Kernmatrix (de Belle et al. 1998). In Halo-FISH-Analysen sind diese DNA-Sequenzen anstatt an der Kernmatrix in der DNA-Halo lokalisiert. T-Zellen in SATB1 knock-out Mäusen arretierten in einer doppelt positiven (CD4+ CD8+) Phase, was innerhalb von 3 Wochen zum Tod der Zellen führte (Yasui et al. 2002).
Zur Zeit wird allgemein davon ausgegangen, dass die Wirkung von SATB1 davon abhängt, mit welchen Transkriptions- oder Silencing-Komplexen das Protein interagiert. SATB1 soll eine Klasse von Transkriptionsregulatoren definieren, die als Andockplattform für eine Reihe von Chromatin Remodelingkomplexen fungieren können und damit Chromatin-Domänen regulieren.
Wenn man die Beobachtungen von Kohwi-Shigematsu und anderen Voruntersuchern mit meinen eigenen Beobachtungen und Berechnungen zusammenführt, lassen sich für SATB-Proteine zwei verschiedene Wirkungsweisen postulieren. Die eine ist mitoseabhängig und mit der Rekrutierung von remodeling-Faktoren oder besser remodeling-Maschinen verbunden hier agieren SATB-Proteine als Silencing-Faktoren - die andere ist mit dem prämitotischen Verpacken des Chromatins zum Chromosom und mit dem postmitotischen Entpacken dieses Chromosoms zu einer transkriptionsfähigen Chromatin-Struktur korreliert.
Sowohl SATB- als auch HMGA-Proteine haben also offensichtlich zwei sehr unterschiedliche Funktionen, die auch während sehr unterschiedlicher Phasen des Zellzyklus zum Tragen kommen. Hier sollen zunächst die Funktionen dieser beiden Proteine bei der Stillegung von Genen bzw. Chromatin-Domänen besprochen werden. Die Funktion dieser beiden Proteine bei der Ver- und Entpackung von DNA und Chromatin werde ich erst im folgenden Teil III ausführlich diskutieren.
SATB-und HMGA-Proteine sind während der Mitose
an der Aktivierung von REMA-Genen beteiligt, die remodeling-Maschinen
für die NG 208 und 150 kodieren.
Chromatin-Domänen, die in der prämitotischen Phase mit der Nukleosomengrösse 208 formatiert wurden, werden nach der Mitose durch Formatierung mit der NG 150 unwiderruflich stillgelegt. Weiter oben hatte ich postuliert, dass an diesem Prozess Silencing-Proteine beteiligt sein müssen, die ähnlich wie ein primäres Transkriptionshormon den Startpunkt der Formatierung festlegen.
Von allen Architekturproteinen, die ich in Teil III noch ausführlich vorstellen und besprechen werde, kommen für diese Funktion nur HMGA- und SATB-Proteine in Frage. HMGA- und SATB-Proteine sind nach dem Ergebnis meiner Berechnungen sowohl an der Aktivierung von REMA-Genen für die Nukleosomengrösse 208 bzw. 150 beteiligt, als auch an der Festlegung der Formatierungs-Startstelle, indem sie als Bestandteil des Aktivierungskomplexes zusammen mit einer remodeling-Maschine für die NG 208 bzw.150 an die korrrelierte REMA-Box binden und die Formatierung der Domäne mit der Nukleosomengrösse 208 (prämitotische Phase) bzw. 150 (postmitotische Phase) induzieren.
Dabei verhält sich das HMGA-Protein wie ein HRPK aus einem Formatierungshormon und einem Rezeptorprotein. Die Synthese von HMGA-Proteinen wird demzufolge durch Formatierungshormone induziert.
SATB-Proteine dagegen verhalten sich wie ein HRPK aus einem primären Transkriptionshormon und einem Rezeptorprotein. SATB-Proteine werden dementsprechend durch primäre Transkriptionshormone aktiviert.
Über die Transkription von REMA-Genen habe ich ausführlich in Teil II diese Arbeit referiert; deshalb sei nur der Vollständigkeit halber erwähnt, dass die zuvor beschriebenen Prozesse Interaktionen zwischen SMARs/REMAKEs der Klasse III und SMARs der Klasse II sowie der Kernmatrix erfordern. Darüber wird in Teil III noch ausführlich gesprochen.
Abgesehen von den erwähnten Besonderheiten verläuft die Inaktivierung einer Chromatin-Domäne durch Markierung mit der NG 208 im prämitotischen Zellzyklus und Formatierung mit der NG150 nach der Zellteilung/Mitose wie die Formatierung einer Domäne mit einer der transkriptionsaktiven Nukleosomengrössen: durch die Aktivierung von Histon-Chaperonen und remodeling-Maschinen werden - wie in Der prämitotische Zellzyklus beschrieben - in den Nukleosomen von Domänen, die inaktiviert werden sollen, ein canonisches H2B- und ein canonisches H3-Monomer durch Monomere aus zellspezifischen paralogen Histon-Varianten und das Standard-H1-Molekül durch eine zellspezifische H1-Variante ersetzt.
 Die stillzulegende Chromatin-Domäne wird unter Mitwirkung eines Silencing-Faktors der HMGA- oder SATB-Gruppe in Verbindung mit einem HRPK im prämitotischen Zellzyklus zunächst mit der NG 208 formatiert. Diese Formatierung führt im postmitotischen Zellzyklus zur Aktivierung einer remodeling-Maschine für die NG 150, die anschliessend zusammen mit einem Aktivierungskomplex an die REMA-Box für die NG 150 im REMAKE der Domäne bindet . Nach der Formatierung mit der NG 150 wird die Domäne durch Bindung von modifiziertem H1 an die Nukleosomen-Cores dauerhaft inaktiviert. Die stillzulegende Chromatin-Domäne wird unter Mitwirkung eines Silencing-Faktors der HMGA- oder SATB-Gruppe in Verbindung mit einem HRPK im prämitotischen Zellzyklus zunächst mit der NG 208 formatiert. Diese Formatierung führt im postmitotischen Zellzyklus zur Aktivierung einer remodeling-Maschine für die NG 150, die anschliessend zusammen mit einem Aktivierungskomplex an die REMA-Box für die NG 150 im REMAKE der Domäne bindet . Nach der Formatierung mit der NG 150 wird die Domäne durch Bindung von modifiziertem H1 an die Nukleosomen-Cores dauerhaft inaktiviert.
Durch zeitgleiche, dynamische Modifizierung der Histone und der DNA werden die Histon-Oktamere auf der DNA derart positioniert, dass die DNA im Anschluß mit der NG 208 formatiert ist. Während des postmitotischen Zellzyklus werden die solcherart markierten Domänen durch Aktivierung spezifischer REMA-Gene für die NG 150 und Assemblierung einer entsprechenden remodeling-Maschine mit der Nukleosomengrösse 150 formatiert und durch Bindung von modifiziertem H1 endgültig stillgelegt.
Die durch Proteine der HMGA- oder SATB- Familie während der Mitose induzierte Inaktivierung von DNA-Abschnitten durch Formatierung mit der Nukleosomengrösse 150 kann auf eine einzelne Domäne beschränkt bleiben, sich aber auch auf eine ganze Transkriptionsgruppe oder gar ein Chromosomenband erstrecken. Da die Organisation der Gene in Subdomänen, Domänen (Chromomere), Transkriptionsgruppen, Chromosomenbändern und Chromosomen nicht planlos erfolgt wie ich hoffe gezeigt zu haben - sondern einer inhärenten Ordnung folgt, wird die Inaktivierung von Chromatin-Domänen, wird die Bildung von kompaktiertem, sogenanntem Heterochromatin mit einiger Sicherheit koordiniert und in größeren Einheiten erfolgen und unter Beteiligung von weiteren Architekturproteinen wie solchen der HMGN-, HMGA- und der ARBP/MeCP2-Gruppe ablaufen. Darüber werde ich unter anderem in Teil III dieser Arbeit sprechen.
Imprinting oder der Kampf der Geschlechter
auf der Ebene der Gene.
Die imprinting genannte allel-spezifische Stillegung eines Gens ist ein Prozess, der nur wenig erforscht ist. Er führt zur Stillegung ausgewählter mütterlicher oder väterlicher Gene157 auf dem jeweiligen Chromosom und damit zu einer spezifischen Ausprägung mütterlicher oder väterlicher Merkmale des Individuums. Hintergrund dieses Mechanismus ist nach allgemeiner Auffassung der Evolutionsbiologen der struggle for life der mütterlichen und väterlichen Gene um die Ressourcen des mütterlichen Organismus.
Imprinting wird interpretiert als Inaktivierung eines mütterlichen oder väterlichen Allels durch Blockierung der Transkription des von der Mutter oder vom Vater geerbten Gens durch Bindung eines spezifischen Proteins an eine sogenannte imprinting-control-region (ICR)158. Eines dieser Proteine CCCTC-binding Faktor oder kurz CTCF genannt - ist nachweislich an der Inaktivierung von Genen des mütterlichen X-Chromosoms beteiligt, indem es an die unmethylierte mütterliche ICR bindet und dadurch die Transkription der mütterlichen Allele blockiert. Auf dem väterlichen Chromosom dagegen verhindert die Methylierung der ICR die Bindung von CTCF, was im Gegenzug zur Aktivierung der väterlichen Allele führt (Bell and Felsenfeld, 2000; Burcin et al.,1997; Hark et al., 2000; Kanduri et al., 2000; Szabo et al., 2000).
Das imprinting-Muster der ererbten mütterlichen und väterlichen Chromosomen wird während der Entwicklung der Keimzellen in der Keimbahn zunächst gelöscht und dann geschlechts-spezifisch wieder aufgebaut - nach meinen Berechnungen zwischen dem 80. und dem 200. Tag der Fetogenese sowie während der Pubertät.
Was weiss man über CTCF?
Das auf Chromosom 16q21q22.3 lokalisierte Gen für CTCF ist ein Mitglied der BORIS- und der CTCF-Gen-Familie. Es besteht vermutlich aus 12 Exons und kodiert (unter anderem) ein Protein aus 727 Aminosäuren. Das Protein soll ubiquitär exprimiert werden und durch Nutzung verschiedener Kombinationen seiner hochkonservierten 11 Zinkfinger-Domänen in der Lage sein, unterschiedliche DNA-Sequenzen und Proteine zu binden.
Abhängig vom genomischen Kontext der Bindestelle kann CTCF einen Komplex binden, der eine Histon-Acetyl-Transferase (HAT) enthält und dann als Transkriptions-Aktivator fungieren oder durch Bindung eines Komplexes, der eine Histon-Deacetylase (HDAC) beinhaltet, als Repressor der Transkription agieren.
Wenn CTCF an ein sogenanntes Insulator-Element bindet, kann es die Interaktion zwischen Enhancer-Elementen und upstream gelegenen Promotoren unterbinden und dadurch die Expression imprinteter Gene regulieren durch synergistisches Zusammenwirken zum Beispiel mit dem Thyroid-Hormonrezeptor soll CTCF aber auch in der Lage sein, die Transkription zu aktivieren.
CTCF scheint also ein sehr vielseitiges Protein sein: ein Protein, das nicht nur am imprinting, also der allel-spezifischen Stillegung von Genen, sondern auch an der Aktivierung von Genen beteiligt ist zwei Funktionen, die nur schwer in einen logischen Zusammenhang zu bringen sind.
Gibt es eine tatsächlich eine
Consensus-Sequenz für die Bindung von CTCF?
Zunächst glaubte man, dass CTCF an keine strikt konservierte Sequenz bindet (Bell and Felsenfeld, 2000; Burcin et al.,1997; Hark et al., 2000; Kanduri et al., 2000) sondern auf Grund seiner varianten Zinkfinger-Motive in der Lage sei, an eine große Zahl nicht konservierter Zielsequenzen zu binden (im Mausgenom hatte man über 10.000 Stellen identifiziert, an die CTCF bindet (Mukhopadhyay et al., 2004)).
Kim et al.159 allerdings gelang es, mittels ChIP-on-chip-Analysen des humanen Genoms ein 20-mer Consensus-Motiv (siehe Abbildung - Quelle: KIM et al.) für die Bindung von CTCF zu etablieren, das allerdings nicht mit allen von der Gruppe identifizierten 13804 CTCF-binding-sites zu korrelieren war. 18 % der in vivo in IMR90-Zellen registrierten CTCF-Bindestellen stimmten nicht mit dem Consensus überein, obwohl auch diese in vitro CTCF zu binden in der Lage waren. Auch ein Teil der bereits früher identifizierten CTCF-Bindestellen konnte nicht mit dem neu gefundenen Consensus in Deckung gebracht werden.
Das Problem scheint mir darin zu bestehen, dass die Gruppe um Kim ausschliesslich mit dem 727aa-CTCF-Protein gearbeitet hat. Dieses Protein ist aber nach meinen Analysen nur eines von 12 Proteinen, die vom CTCF-Gen auf Chromosom 16q21-q22.3 in drei Phasen zwischen dem 80. und dem 160., dem 160. und dem 200. Tag und während der Pubertät exprimiert werden.
Die zwölf durch das CTCF-Gen kodierten Proteine, drei davon hier in ENSEMBLE dokumentiert160, werden nach meinen Berechnungen zu verschiedenen Zeiten und in verschiedenen Zellen exprimiert die translatierten Proteine unterscheiden sich nicht nur in der Zahl ihrer Aminosäurereste, sondern auch in der Zahl ihrer Zinkfinger-Domänen.
Nutzen CTCF-Proteine einen Zinkfinger-Code, um ihre Bindestellen zu identifizieren?
Man weiss, dass CTCF unterschiedliche Kombinationen seiner Zinkfinger-Domänen zur Bindung unterschiedlicher DNA-Zielsequenzen nutzt. Die unterschiedlichen Nutzung von Zinkfinger-Domänen könnte also einen Code darstellen, mit dessen Hilfe die verschiedenen CTCF-Proteine ihre unterschiedlich modifizierten Bindestellen identifizieren. Dann gäbe es zwar nach wie vor nur 12 CTCF-Proteine, aber eine wesentlich grössere Zahl von Bindemotiven, die sich allerdings alle sehr ähnlich sehen müssten. Aus Gründen, die ich nun erläutern werde, nehme ich an, dass die Zahl der Zinkfinger-Codes der zwölf postulierten CTCF-Proteine mit der Anzahl der von mir postulierten sekundären Transkriptionshormone (das sind 52) übereinstimmt.
Im menschlichen Genom gibt es nach meinen Berechnungen rund 110000 (einhundertundzehntausend) SMARs der Klasse III davon sind 26500 REMAKEs161, also SMARs der Klasse III, die REMA-Boxen enthalten, an welche ATP-abhängige remodeling-Komplexe und verschiedene Architekturproteine binden. Diese 26500 REMA-Boxen enthalten nach meinen Hochrechnungen jeweils eine Bindestelle für eines der zwölf Proteine der CTCF-Familie.
Nach meinen Berechnungen entfallen von den oben genannten insgesamt 26500 potentiellen Bindestellen für Proteine der CTCF-Familie ca. 10500 Bindestellen auf das Protein CTCF-001, also die 727aa Variante. Auf die 399aa-Variante entfallen ca. 1900 und auf die 443aa-Form ca. 3100 CTCF-Bindestellen. Der Rest von insgesamt 26500 Bindestellen, also 11000 Bindestellen verteilt sich auf die restlichen 9 (neun) noch unidentifizierten CTCF-Proteine. Damit stimmen meine Zahlen sehr gut mit den von Kim et al. ermittelten überein und bieten gleichzeitig eine Erklärung für die Tatsache, dass die von ihm gefundene Consensus-Sequenz nicht mit allen CTCF-Bindestellen in Übereinstimmung zu bringen war.
CTCF-Proteine binden an Insulator-Regionen das sind SMARS
der Klasse III, die REMA-Boxen enthalten.
Die in der Literatur heute allgemein als Insulatoren bezeichneten DNA-Regionen sind das habe ich weiter oben schon einmal gesagt mit einiger Sicherheit mit den SMARs der Klasse III identisch, die abhängig vom Zellzyklus sowohl REMAKEs als auch Subdomänen-SMARs darstellen können. Als REMAKEs dienen sie während der Mitose als Andockstelle für die diversen remodeling-Maschinen, welche über die Formatierung der Domäne die entwicklungs- und zellspezifische Aktivierung oder Inaktivierung von Genen in dieser Domäne steuern als Subdomänen-SMARs binden sie in der Interphase solche Proteine, welche die Ausbildung einer spezifischen Subdomänen-Schleife und der primären und sekundären Transkriptionsschleife induzieren, die aus dieser Subdomänen-Schleife entwickelt werden. Darüber werden wir später noch mehr lesen.
Das Protein, welches die Funktion eines REMAKEs als Andockstelle für Remodeling-Maschinen im prämitotischen Zellzyklus determiniert, ist in der frühen Phase der Ontogenese (bis etwa zum 80. Tag) ein Protein der HMGA-Gruppe, in der Zeit danach ein Protein der HMGA- und/oder154 der SATB-Gruppe. CTCF-Proteine binden wahrscheinlich erst ab dem 80 Tag an REMAKEs - und das wahrscheinlich nur an solche REMAKEs, die in einer REMA-Box 208 bzw. 150 ein HMGA- und ein SATB-Protein gebunden haben, also an REMAKEs, die Transkriptionsgruppen determinieren.
SATB und CTCF sind Interaktionspartner.
Nach dem Ergebnis meiner Untersuchungen und Berechnungen sind SATB-Proteine und Proteine der CTCF-Familie Interaktionspartner: diesen Zusammenhang sehe ich auch durch Untersuchungsergebnisse Anderer bestätigt, in denen SATB-Proteine ebenso wie CTCF-Proteine mit der Rekrutierung von remodeling-Faktoren bzw. maschinen in Verbindung gebracht werden.
Diese während einer Mitose von SATB rekrutierten remodeling-Faktoren sind wie ich weiter oben ausgeführt habe an der Formatierung einer Domäne mit der NG 208 bzw. 150 beteiligt. Diese Formatierung kann sich aber - nach allem, was ich bisher über die Mechanismen und Prozesse gesagt habe, die dem remodeling von Chromatin-Domänen zugrundeliegen nur auf beide Allele eines definierten Gens erstrecken162. Daraus ergibt sich die logische Schlussfolgerung, dass imprinting nicht auf der Stillegung von Transkriptionsgruppen oder Domänen durch Formatierung dieser DNA-Abschnitte mit der NG 208/150 beruhen kann, da imprinting per definitionem die Inaktivierung eines maternalen oder paternalen Gens bedeutet.
Die Funktion von CTCF beruht nicht auf der Inhibierung
der Gen-Aktivierung, sondern auf der Inhibierung der Gen-Inaktivierung.
Wenn man allerdings annimmt und dafür gibt es mehr als deutliche Hinweise dass Imprinting sehr wohl mit der Stillegung von Genen durch Formatierung mit der NG 208/150 verbunden ist, dann muss man zu dem ebenso logischen Schluss kommen, dass Imprinting nicht auf der Inaktivierung von Genen einer Transkriptionsgruppe, sondern auf der Repression ihrer Inaktivierung beruhen muss. Die Funktion von CTCF verkehrt sich dann von einer Inhibierung der Aktivierung in eine Inhibierung der Inaktivierung.
Imprinting ist nach dem Ergebnis meiner Untersuchungen und Berechnungen aufs Engste mit der globalen, also beide Allele einschliessenden Stillegung von Genen durch Formatierung von Chromatin-Domänen mit der Nukleosomengrösse 150 gekoppelt. Diese Stillegung wird wie ich oben ausgeführt habe - von einem spezifischen HMGA- bzw. SATB-Protein vermittelt, das als Bestandteil des Transkriptionskomplexes zunächst an die ASP und SP von REMA-Genen für die NG 208 bzw. 150 und anschliessend zusammen mit dem Aktivierungskomplex an die REMA-Box für die gleiche Nukleosomengrösse bindet. Dieser dynamische Prozess läuft - wie weiter oben beschrieben - in Interaktion mit der Kernmatrix ab und führt über die Aquirierung einer remodeling-Maschine für die Nukleosomengrösse 208 bzw.150 zur Stillegung der Domäne.
Die Bindung von CTCF an eine spezifisch modifizierte REMA-Box 208 bzw. 150 in der REMAKE-Region eines DNA-Doppelstrangs verhindert das Andocken von remodeling-Maschinen an diese Box, blockiert damit die Formatierung dieser Domäne mit der NG 208 bzw.150 und unterbindet folglich ihre Stillegung. Die Bindung von CTCF an die REMA-Box 208/150 im REMAKE eines DNA-Doppelstrangs was die Inaktivierung der korrelierten Domäne dieses Doppelstrangs verhindert - ist also mit der Stillegung der homologen Chromatin-Domäne des anderen Doppelstrangs verknüpft. Insofern ist CTCF sowohl am Imprinting, der selektiven, allel-spezifischen Inaktivierung als auch an der Aktivierung von Genen beteiligt.
Durch das hier Vorgetragene lassen sich zumindest einige der beschriebenen Phänomene im Zusammenhang mit CTCF erklären: so die große Zahl von potentiellen Bindungsstellen (über 10.000 im Mausgenom)163 die Blockierung der Enhancer-Wirkung von DNA-Elementen und die damit verknüpfte Funktion als Isolator oder die Beziehung von CTCF zur Aktivierung oder Rekrutierung von Histonazetylasen oder deazetylasen und die Beziehung zu Promotoren und Hormon-Rezeptorproteinkomplexen.
Hat der epigenetische RESET etwas mit dem Imprinting zu tun?
Eingangs dieser Arbeit habe ich postuliert, dass die Formatierungskaskade von einem epigenetischen Reset ausgehen muss, den ich etwa für die 80. Stunde p.c. festgelegt habe. Dieser Reset ist so habe ich vermutet mit wechselseitigen, vom Cytosplasma der Zygote initiierten Demethylierungen der vom Vater bzw. der Mutter ererbten Genome verbunden. Nach welchen Regeln oder Prinzipien die Auswahl der Gene erfolgt, die imprinted werden, habe ich nicht untersucht. Ich werde das zu einem späteren Zeitpunkt eventuell nachholen. Ich vermute aber, dass das Imprinting-Muster durch Methylierungen/Demethylierungen anlässlich dieses von mir postulierten epigenetischen Resets festgelegt wird.
RNA-Interferenz und Sirtuine.
Da chromatin-remodeling oder Formatierung der DNA auf der Domänenebene stattfindet, würde die Formatierung einer Domäne mit der NG 150 und der Bindung eines H1-Histons bedeuten, dass alle Gene dieser Domäne stillgelegt sind. Da die Formatierung mit der NG 150 vermutlich nicht nur mit der Bindung von H1, sondern auch mit der Bildung übergeordneter Strukturen (Solenoid, Chromomer) gekoppelt ist, ist die Stillegung einer Domäne und der Gene darin durch Formatierung mit der NG 150 demnach mit einiger Sicherheit irreversibel.
Um in einer Domäne, die mit einer transkriptionsaktiven NG formatiert ist, einzelne Gene (vorübergehend) stillzulegen oder abzuschalten, greift die Zelle deshalb auf verschiedene Mechanismen zurück, die zur Zeit zum Teil intensiv diskutiert und untersucht werden. Ein solcher Mechanismus ist die bereits erwähnte RNA-Interferenz, die allerdings auf der posttranskriptionellen Ebene ansetzt. Ein anderer könnte die temporäre und reversible Einwirkung spezialisierter Proteine auf die tails von Histonen sein, ein Prozess, der zu einer verdichteten, kompaktierten und damit unzugänglichen Chromatin-Struktur führen könnte. Hier sind die sogenannten Sirtuine in der Diskussion, Histon-Deacetylasen der Klasse III, die durch Deacetylierung spezifischer Lysinreste eine Kompaktierung des Chromatins bewirken können.
|



{kind=link}
{kind=link}