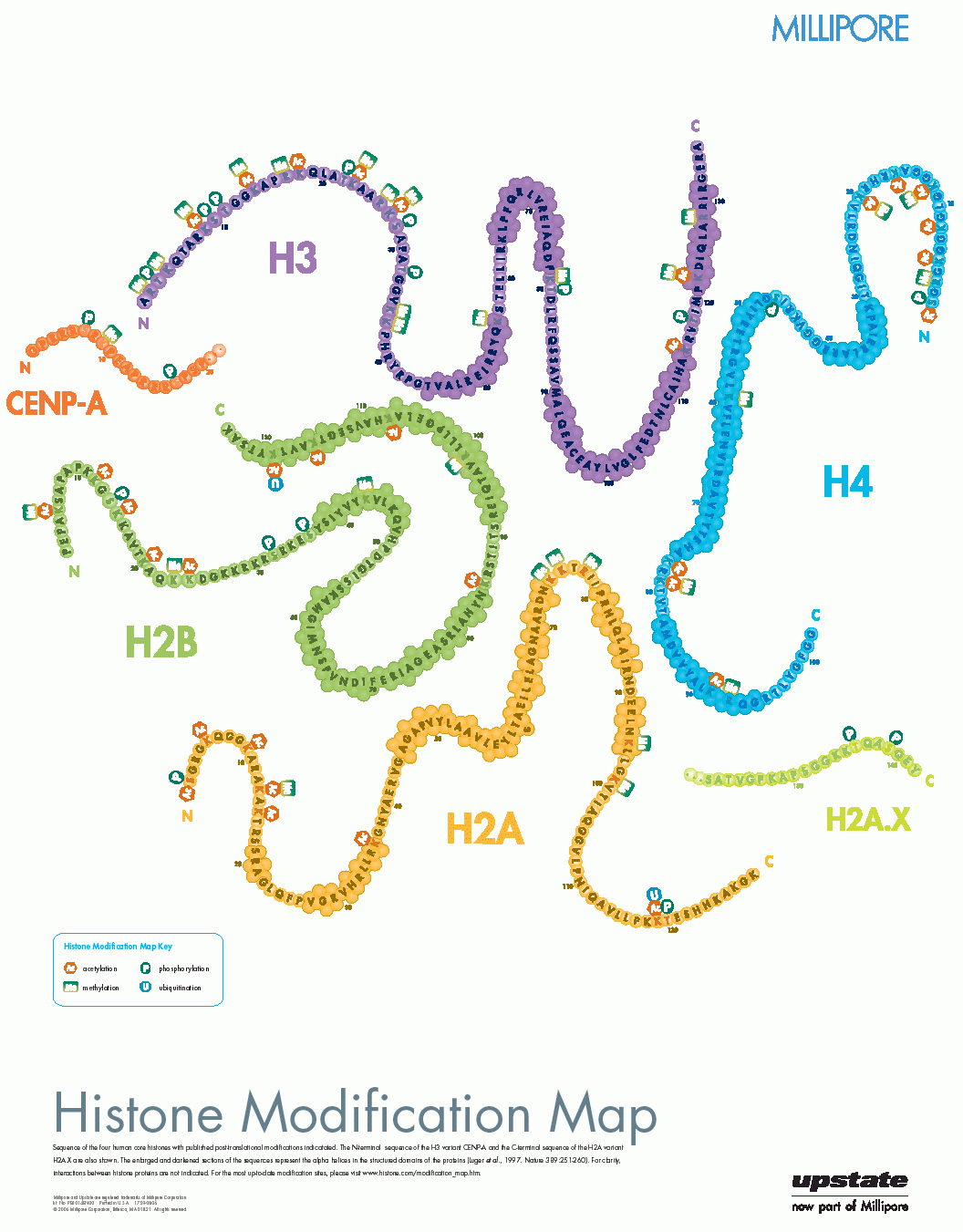|
|
 |
|
|
|
Wolfgang Marks: Die Formatierte DNA
|
|
|
Der Transkription erster Schritt: Der Weg zum Primärtranskript.
Der Histon-Code: in Wahrheit ein Histon-DNA-Code?
Der Autor dieses Textes ist sich sicher, dass der von Strahl und Allis im Jahre 2000 postulierte Histon-Code9 in Wahrheit ein Histon-DNA-Code ist, ein Code, der wie von mir bereits 1991 formuliert - von der Zelle bei jedem Differenzierungs- oder Proliferationsschritt genutzt wird, um die DNA ähnlich wie eine Computer-Festplatte zu formatieren sie also mittels eines Codes von chemischen Modifikationen an DNA-Nukleotiden und Histonen in definierte Segmente oder Abschnitte einzuteilen, die mit Nukleosomen einer distinkten Grösse in Phase besetzt werden und es der Zelle so erlaubt, im weiteren Fortgang der Ontogenese die spezifisch formatierten Abschnitte und damit ganz bestimmte Sequenzen der DNA gezielt zu aktivieren oder zu deaktivieren.
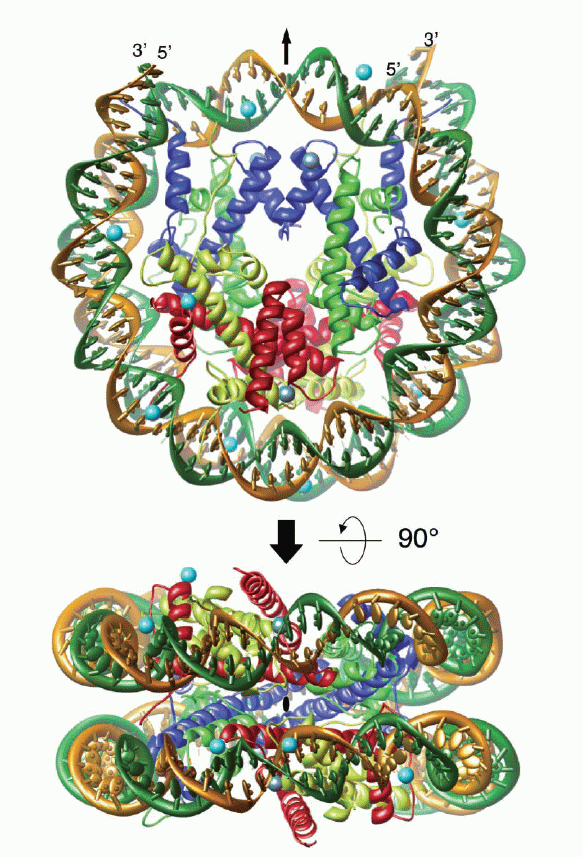
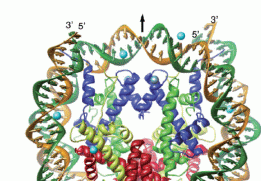 Kristall-Struktur-Modell (ribbon-model) eines Nukleosomenkerns bestehend aus einem Histon-Oktamer, das von DNA umschlossen ist, in verschiedenen Ansichten. Abbildung mit freundlicher Genehmigung entnommen aus: Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle; Authors: Yasuo Tsunaka, Naoko Kajimura, Shinichi Tate and Kosuke Morikawa, 34243434; Nucleic Acids Research, 2005, Vol. 33, No. 10 Kristall-Struktur-Modell (ribbon-model) eines Nukleosomenkerns bestehend aus einem Histon-Oktamer, das von DNA umschlossen ist, in verschiedenen Ansichten. Abbildung mit freundlicher Genehmigung entnommen aus: Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle; Authors: Yasuo Tsunaka, Naoko Kajimura, Shinichi Tate and Kosuke Morikawa, 34243434; Nucleic Acids Research, 2005, Vol. 33, No. 10
Was sind Nukleosomen?
Der Nukeosomenkern (das Core) besteht aus einem Komplex aus acht Proteinen, dem sogenannten Histon-Oktamer, das von 146 Basenpaaren10 DNA umschlossen ist. Wir wissen, dass die Anzahl der Basenpaare der linker-DNA zwischen den Nukleosomenkernen schwankt. Während die einen für die linker-DNA Werte zwischen 10 und 50 bp angeben, sprechen andere von 10 bis 90 oder sogar von 10 bis 114 bp. Für die Nukleosomengröße11, also für die Summe aus den Basenpaaren, die an das Oktamer gebunden sind und den Basenpaaren der sogenannten linker-DNA, werden dementsprechend in der Literatur die unterschiedlichsten Werte genannt. Die Spanne reicht von 154 bis ca. 260. (Nach meinen Berechnungen beträgt die minimale Nukleosomengröße im menschlichen Genom 150 bp, die maximale derzeit 260 bp - dazu später mehr).
DNA, die transkribiert werden soll, muss für Transkriptionsfaktoren und regulatoren zugänglich sein. Darüber besteht weitestgehend Einigkeit. Einig ist man sich auch darüber, dass DNA, die um einen Komplex, ein Core aus Histon-Oktameren gewunden ist, nicht abgelesen werden kann. Ob und wie oder welche Histone sich während der Transkription von der DNA lösen, war für das hier zur Debatte stehende mathematische Denkmodell und das darauf basierende Programm zunächst nicht relevant. Auch die verschiedenen Modifikationen der Histone, ihrer tails oder der DNA selbst waren für den Autor nur insoweit von Interesse, als diese Modifikationen nach Annahme des Autors Auswirkungen auf die Bestückung der DNA mit Nukleosomen einer spezifischen Grösse und und einer bestimmten inneren Struktur haben.
Wie bereits erwähnt, ging der Autor schon 1989/90 bei seinen ersten Überlegungen davon aus, dass in den Sequenzen der DNA eine Ordnung existiert, die zwar die Funktion der DNA als RNA-Chiffre begründet, aber weit darüber hinausgehend eine immanente, mathematisch erfass- und beschreibbare Überordnung in der Anordnung spezifischer Sequenzmuster einschließt. Denn Proteine, die als Aktivatoren, Repressoren, Modulatoren oder Enhancer an die DNA binden, tun dies ja nicht zufällig, sondern an Stellen, die dafür im Wortsinne prädestiniert sind. Auch die Position der Histon-Oktamere auf oder an der DNA ist ja - wie später noch diskutiert werden wird nicht zufällig, sondern erfolgt offensichtlich nach bestimmten Mustern oder Regeln, die einen direkten Einfluss auf die Genexpression haben. Die in der Literatur beschriebenen Histon- und DNA-Modifikationen können also wohl kaum zufällig sein, sondern müssen sämtlich in einem logischen Zusammenhang stehen.
Der zunächst vermutete Zusammenhang Euchromatin gleich aktive Gene Heterochromatin gleich inaktive Gene hat sich rasch als unzutreffend erwiesen. Schon in den 90er Jahren haben Untersuchungen über die Zugänglichkeit von Genen für den Transkriptionskomplex gezeigt, daß auch in DNA, die offensichtlich als Euchromatin vorliegt, die Transkription von Genen inhibiert sein kann. Es konnte nachgewiesen werden, dass diese Repression durch spezifisch positionierte Nukleosomen vermittelt wird, die bestimmte DNA-Regionen besetzen. Auf der anderen Seite haben genetische und biochemische Analysen aber auch schon früh ergeben, daß die gleiche (Eu)-Chromatinstruktur eine ebenso entscheidende Rolle bei der Aktivierung der Transkription spielt und die Transkriptions-Initiation vereinfachen kann (Wallrath et al., 1994; Wolffe, 1994).
Genexpression und regulation ist mit der Anordnung
der Nukleosomen auf der DNA verknüpft.
Die Position von Nukleosomen12 in einer Promotor-Region hat einen entscheidenden Einfluß auf die Genregulation. So werden die Promotoren des 'mouse mammary tumor virus' (MMTV) und des Hefe PHO5 Promotors durch spezifisch positionierte Nukleosomen reguliert (Bresnick et al., 1992; Lee und Archer, 1994; Straka und Hörz, 1991). Die Analyse der Vorgänge bei der Regulation der entsprechenden Gene führte zu dem Schluß, dass die spezifische Anordnung der Nukleosomen am Promotor entscheidend ist für die Regulation der Transkription.
In dieses Bild passt auch die Beobachtung, dass spezifisch positionierte Nukleosomen, die zwischen dem Promotor und stromaufwärts liegenden aktivierenden Sequenzen lokalisiert sind, für die Bildung des Transkriptions-Initiationskomplexes notwendig sind. Derartig positionierte Nukleosomen vereinfachen die Interaktion von Proteinen mit entfernt liegenden DNA-Bindungssequenzen. Dies wurde z.B. am Drosophila hsp26-Promotor und am Xenopus Vitellogenin B1-Promotor beobachtet (Schild et al., 1993; Lu et al., 1993; Lu et al., 1995).
Dass die Anordnung der Nukleosomen auf der DNA nicht statisch ist, sondern in Abhängigkeit von der Gensequenz und dem Aktivitätszustand des betreffenden Gens Unterschiede in der Länge der linker-DNA aufweist, ist ein weiterer Beleg dafür, dass das Schicksal eines Gens offensichtlich mit der Nukleosomengrösse verknüpft ist13.
Die Ermittlung von Nukleosomenpositionen mittels ChIP -
brauchbar oder unbrauchbar?
In jüngster Zeit haben mehrere Gruppen von Untersuchern es unternommen, große Teile des Genoms von saccharomyces cerevisiae (Bäckerhefe) daraufhin zu analysieren, wie die DNA dieser Schlauchpilze in Nukleosomen organisiert ist. Für diese Untersuchungen wurden neue Techniken entwickelt, mit denen große Teile von Genomen oder auch ganze Genome mit Hilfe spezifischer Antikörper gegen definierte Modifikationen an bestimmten Histonen auf das Vorhandensein von Nukleosomen überprüft werden können. Sowohl die sogenannte ChIP14 on chip Methode als auch die ChIP-Seq- Methode bergen aber Fehlermöglichkeiten in sich, die zum einen in der Komplexität der Verfahren selbst zu suchen sind, zum anderen in der Spezifität der ausgewählten Antikörper und der Heterogenität der verwendeten Zellkulturen. Da die Zellen einer lebenden Kultur sich in verschiedenen Stadien der Entwicklung und/oder des Zellzyklus befinden, wird eine ChIP-Seq- Analyse von Nukleosomenpositionen in diesen Zellen immer screen-shots liefern, die durch Überlagerungseffekte überall dort unscharf werden, wo Nukleosomenpositionen sich aufgrund unterschiedlicher Differenzierungs- oder Zyklusstadien der jeweiligen Zellen verändert haben. Gleiches gilt für die Ermittlung von Nukleosomenpositionen mit Hilfe spezifischer Antikörper gegen bestimmte Histone (z.B. H2A.Z) oder gegen spezifische Modifikationen an bestimmten Histonen (zum Beispiel an H3 oder H4).
Da die verwendeten Antikörper sich immer nur gegen eine bestimmte Modifikation an einem bestimmten Histon oder gegen eine definierte (kleine) Zahl von solchen Modifikationen richten, können nur solche Nukleosomen entdeckt werden, die auch ein Histon mit diesen Modifikationen enthalten. Da wie ich in dieser Arbeit unter anderem zeigen will, aber nicht eine einzelne Modifikation oder deren drei oder vier an einem einzelnen Histon, sondern eine ganz bestimmte Kombination von Modifikationen an unterschiedlichen Histonen und an der DNA (der später von mir so genannte genomische Schlüssel, der Histon-DNA-Code) zu einer zellspezifischen Positionierung von Nukleosomen auf der DNA führt, sind die Ergebnisse der beiden zuvor genannten Analysemethoden nach meinem Dafürhalten nur bedingt brauchbar.
Gleiches gilt für die unter der Adresse: http://h2az.atlas.bx.psu.edu/ veröffentlichte Nukleosomenkarte des Genoms von Saccharomyces Cerevisiae: auch diese Karte sagt m.E. nichts über die tatsächliche Position eines Nukleosoms in einem bestimmten Stadium der Differenzierung oder des Zellzyklus von Sacch. C. aus, weil es keine Zellkultur gibt, deren Zellen sich alle in der gleichen Arbeitsphase oder im gleichen Stadium des Zellzyklus befinden15. Dies - und die zuverlässige Fixierung dieses Zustandes wäre aber die grundlegende Voraussetzung für eine ChIP-Analyse gleich welcher Art, die nicht von Überlagerungseffekten geprägt und dominiert sein soll.
Ich sage dies, obwohl ChIP-on-Chip als auch ChIP-Seq Ergebnisse liefern, die meine Theorie in vielen Punkten stützen: so ist zum Beispiel die Tatsache, dass Promotor-Regionen und poly-A-Regionen grundsätzlich nicht von Nukleosomen besetzt werden16 oder dass im Transkriptionsbereich von Genen ein regelmässiges spacing vorherrscht, ein sicherer Hinweis darauf, dass Promotoren, Terminatoren und andere essentielle Genombereiche in ein System eingebunden sein müssen, das die Positionierung von Nukleosomen genomweit steuert.
In den letzten zehn Jahren ist eine Vielzahl von Beobachtungen gemacht worden, welche die herausragende und spezifische Funktion der Histone bei der Kontrolle der Genaktivität unterstreichen (Grunstein, 1990; Grunstein, 1997). Eliminierung der Histone durch gezielte Ausschaltung einzelner Histon-Gene führt zur Aktivierung von Genen, die unter sonst gleichen Bedingungen inaktiv wären (Han und Grunstein, 1988). Punktmutationen innerhalb der N-terminalen Domänen der Core-Histone, welche die Basizität dieser Domänen verringern (Lysin wird durch Glutamin ersetzt) ermöglichen die Transkription von Genen und zwar unabhängig von spezifischen Cofaktoren (Zhang et al., 1998a). Nach Beobachtungen von Kornberg und Lorch (1999) sollen sowohl Wechselwirkungen der zentralen Oktamerregion (histon-fold-motif) mit der DNA als auch Interaktionen der sogenannten tails also der aus dem Kern herausragenden N- und C-terminalen Domänen der Kern-Histone zur transkriptionellen Repression beitragen.
Die DNA ist abschnittsweise in Nukleosomen17 der gleichen Größe in Phase organisiert.
Wenn man die Vielzahl der Untersuchungen und Beobachtungen im Zusammenhang mit Histon-Modifikationen zusammenfasst, fällt auf, dass ein expliziter kausaler Zusammenhang zwischen spezifischen Modifikationen von Histonen die zur Formulierung der Histon-Code-Hypothese führten - und spezifischen Modifikationen der DNA bisher nicht hergestellt wurde. Dabei läge es nahe, einen solchen kausalen Zusammenhang anzunehmen, also davon auszugehen, dass die Modifikationen der DNA die sich nicht allein auf die Methylierung bestimmter Nukleotide beschränken - und die von Histon-Proteinen, die zu einer spezifischen Anordnung der Nukleosomen auf der DNA führen, ein Kopplungsschema aufweisen.
Wenn es dieses Kopplungsschema gibt (was ich hoffe, in dieser Arbeit zeigen zu können) wäre der von Strahl und Allis postulierte Histon-Code in Wahrheit (wie ich es bereits 1991 formuliert habe) ein Histon-DNA-Code - ein Code, der die spezifische Modifikation von Histonen und die definierter Nukleotide der DNA so miteinander verknüpft, dass sie zu einer regelmäßigen (phasengetreuen) Anordnung von Histon-Oktameren auf der DNA führen.
Dies ist die These aus meiner Arbeit von 1991, die ich in dieser Arbeit versuchen will zu beweisen:
- In einer eukariotischen Zelle, die sich in der Interphase befindet, sind definierte Nukleotide der DNA in umschriebenen Abschnitten18 des Genoms gleichartig modifiziert und mit Histon-Oktameren verbunden, die aus gleichartig modifizierten Histonen bestehen. Die DNA ist dadurch abschnittsweise in Nukleosomen19 der gleichen Größe in Phase organisiert. Die Modifizierung von Aminosäureresten der Histone in diesen Abschnitten sind ebenso wie die mit ihnen gekoppelten Modifikationen der DNA einheitlich und für die jeweilige Zelle und das Stadium ihrer Differenzierung spezifisch.
|
|
|
|
|
Der Histon-DNA-Code: Genomische Schlüssel definieren
die Nukleosomenstruktur der DNA.
In der Literatur der letzten Jahre sind ein Reihe von Modifikationen sowohl der DNA als auch der Histone (bzw. deren tails) beschrieben worden, die zumindest eins erkennbar werden lassen: Methylierung von DNA, insbesondere von Guanin und Cytosin führt offensichtlich zur Inhibierung der Transkription, im Gegensatz dazu ist die Azetylierung definierter Aminosäuren von Histonen anscheinend eine conditio sine qua non der Genexpression. Allerdings ist auch schon jetzt erkennbar, dass die möglichen Modifikationen der DNA und der Histone damit nicht annähernd erschöpfend abgehandelt sind.
Wie zuvor erwähnt, hat vanHolde bereits 1988 nachgewiesen, dass Histone das Ziel verschiedener posttranslationaler Modifizierungen sind, wie zum Beipiel Acetylierung, Methylierung, Phosphorylierung und Ubiquitinierung20. Enzyme/Proteine, die diese chemischen Veränderungen hätten bewirken können, waren lange Zeit unbekannt. Erst eine Gruppe um den bereits erwähnten D. Allis konnte Anfang 2000 (Strahl, B.D., and Allis, D. (2000) The language of covalent histone modifications. Nature 403, 41-45. ) ein Protein (CGN5) nachweisen, das Histone acetylierte und ein bereits bekannter21 Transkriptionsregulator war.
Mittlerweile wurden eine ganze Reihe von Proteinen identifiziert, die spezifisch Histone (und DNA-Nukleotide) modifizieren: Lysin-spezifische Histon-Methyltransferasen (HMT) zum Beispiel methylieren definierte, phylogenetisch hochkonservierte Lysine oder Arginine in der NH2-terminalen Region (den tails) von H3 und H4. Dabei ist diese Methylierung distinkter Lysine eng verknüpft mit dem Ablauf bestimmter biologischer Prozesse: die Methylierung von Lysin 9 in H3 (H3K9) von Tetrahymena spec. führt zum Silencing bestimmter Genomabschnitte, während die Methylierung von Lysin 4 (H3K4) mit der Aktivierung der Transkription verbunden ist.
Neuere Untersuchungen zeigen, dass Protein-Methylierungen an der Steuerung zahlreicher biologischer Prozesse beteiligt sind. Dazu gehören RNA-Prozessierung, Transkription, Reparatur von DNA-Schäden sowie die Signaltransduktion. Auch die Differenzierung einer Zelle soll u.a. durch Protein-Methylierung beeinflusst werden (Übersicht in Lee et al., 2005).
Erst in jüngster Zeit wurden verschiedene Protein-Methyltransferasen identifiziert, die Arginin-, Lysin-, Histidin-, Prolin- und Carboxylreste von Aminosäuren methylieren. Solche Methylierungen galten noch bis zum Jahr 2004 für irreversibel und wurden - wie weiter oben schon gesagt mit dem Silencing, also dem Abschalten von Genen in Verbindung gebracht. Die Entdeckung einer Lysin-spezifischen Demethylase (LSD1) brachte dieses Dogma zu Fall und sorgte gleichzeitig für neue Einblicke in die Funktion der Protein-Methylierung (Shi et al., 2004).
Während die Methylierung von Argininresten an zahlreichen RNA-Bindeproteinen und Histonen nachgewiesen werden konnte, findet die gleichartige Modifikation von Lysinen bevorzugt an den amino- und carboxyterminalen tails von Histonen statt. Ebenso wie die Acetylierung kann die Histon-Methylierung die Wechselwirkungen zwischen der DNA und Chromatin-assoziierten Proteinen modulieren, was zu veränderten nukleosomalen Strukturen und Funktionen führt und in den verschiedensten biologischen Wirkungen resultiert (Rice & Allis, 2001).
Eine Arbeitsgruppe um Frank Sauer vom Zentrum für Molekulare Biologie der Universität Heidelberg (ZMBH) hat in den vergangenen Jahren zahlreiche HMTs charakterisiert und nachgewiesen, dass Mutationen in Ash1 einer Histon-Methyltransferase, die spezifisch Lysinreste in H3 und H4 methyliert in Drosophila homozygot letal sind. (Beisel, C., Imhof, A., Greene, J., Kremmer, E., and Sauer, F. (2002) Histone methylation by the Drosophila epigenetic transcriptional regulator Ash1. Nature 419 857-862.)
Wieviele Schlüssel braucht der Mensch?
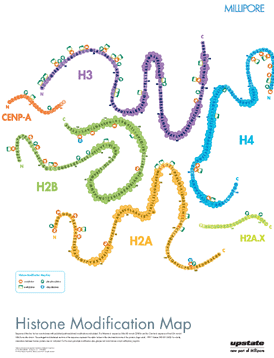
Da die Zahl der entdeckten Histon-Modifikationen ebenso ständig wie rasant wächst, ist es schwierig, einen aktuellen Überblick zu bekommen, da die relevanten Informationen zudem über das www und über eine Reihe verschiedener Fachzeitschriften verstreut sind. Eine schon etwas ältere22 , aber immer noch informative Darstellung in Form eines Posters findet sich auf der (kommerziellen) Webseite der Firma Millipore23:
|
|
|
|

|
|
Eine systematische, sehr gute Übersicht über den (wahrscheinlich) aktuellen Stand des Wissens auf dem Gebiet der Histon-Modifikationen mit Quellenangabe findet sich auf der (ebenfalls kommerziellen) Webseite24 der Firma ABCAM. Die obige Abbildung ist ein Ausschnitt aus einem pdf-Poster, das von dieser Seite heruntergeladen werden kann.
|
|
|
Erst in neuerer Zeit wurden von allen Histonen bislang ausser von H4 zahlreiche Sequenzvarianten entdeckt, also Histon-Proteine, deren Aminosäure-Sequenz nicht mit denen der bekannten Standard-Histone übereinstimmt. (Gilbert et al., 2005). Der Austausch eines Standard-Histons durch eine seiner Varianten soll zu den unterschiedlichsten Phänomenen führen: eine in Säugetierzellen häufig auftretende Variation des Histons H3 z.B. ist das Protein CENP-A. Die Konformation von Nukleosomen, die CENP-A anstelle von H3 enthalten, scheint stabiler zu sein als die Standardkonfiguration und wird u.a. mit der Bildung eines funktionellen Kinetochors in Zusammenhang gebracht. (Black et al., 2004). Das Protein H2A.Z ist eine essentielle Histonvariante von H2A, die vor allem im pericentromerischen Heterochromatin beobachtet wurde. Auch Nukleosomen mit H2A.Z zeigen eine veränderte Konformation, was in bestimmten Chromatinsegmenten zu einer stärkeren intramolekularen Faltung und zur Bildung von hoch kompakten Chromatinstrukturen führt (Fan et al., 2002). Die H2A-Variante MacroH2A (mH2A) wurde bislang nur im inaktiven X-Chromosom entdeckt. Man vermutet, dass sie zur kompakteren Struktur des Xi und Stillegung der Gene beiträgt (Costanzi and Pehrson, 1998; Gilbert and Ramsahoye, 2005). Die H3 Variante H3.3 schließlich wird ausschließlich in transkriptionell aktivem Chromatin gefunden und scheint zu einer offeneren Chromatinstruktur zu führen. (McKittrick et al., 2004).
Von H1 sind zur Zeit (Juli 2009) sechs Varianten bekannt25 . Sie werden beginnend mit H1(0), dem Standard-Histon, fortlaufend numeriert. Zur Zeit ist man bei H1.5 angelangt. Eine Besonderheit stellt anscheinend H1.T dar, das man bislang nur in testis (Hoden) von Maus, Ratte, Affe und Mensch gefunden hat.
Eine Beobachtung im Zusammenhang mit Histon-Varianten ist im Rahmen dieser Arbeit von besonderem Interesse: wie Henikoff et al. festgestellt haben, werden bestimmte Chromatinbereiche dadurch markiert26 , dass die Standard-Core-Histone durch bestimmte Histon-Varianten ersetzt werden. Dieser Austausch von canonischen Histonen gegen spezifische Histon-Varianten wird uns später27 noch besonders interessieren.
Es ist schwierig, wenn nicht gar unmöglich, die vielen Beobachtungen 28 (ich habe bei weitem nicht alle hier erwähnt), die im Zusammenhang mit Modifikationen der Standard-Histone und dem Ersatz dieser Histone durch Histon-Varianten gemacht wurden, in einen sinnvollen Zusammenhang zu bringen, zumal sie sich im einen oder anderen Fall widersprechen. Wenn ich dies trotzdem in dieser Arbeit versuche, bin ich mir bewußt, dass dies nur ein erster sicher nicht vollkommener Versuch sein kann, Ordnung in das System « Chromatin-remodeling » zu bringen, in ein System, das sich mehr und mehr als Schlüssel zum Verständnis der Mechanismen der Genexpression und der epigenetischen Vererbung von Chromatin-Strukturen erweist.
Standard-Histone29 , Histon-Varianten und Modifikationen der DNA.
Modifikationen der vier Nukleotide der DNA sind wenige beschrieben. Methylierung vor allem, aber auch Azetylierung, Phosphorylierung, Ubiquitinisierung, Ribosylierung und andere Modifikationen sind in der Diskussion. Ich habe auf der Basis der bekannten Modifikationen und unter Vorgabe verschiedener Prämissen (definierte Zahl von möglichen Nukleosomengrössen postulierte Zahl von remodeling-Maschinen u.a.) mit Hilfe von Hochrechnungen versucht, die Zahl der möglichen Veränderungen an den Standard-Core-Histonen, an den DNA-Nukleotiden und am Histon H1 (Standard-Protein) annäherungsweise zu berechnen. Diese Berechnungen haben zu folgendem Ergebnis geführt:
|
|
|
|
|
Zahl der möglichen bzw. wahrscheinlichen Modifikationen an den Standard-Histonen und der DNA:
|
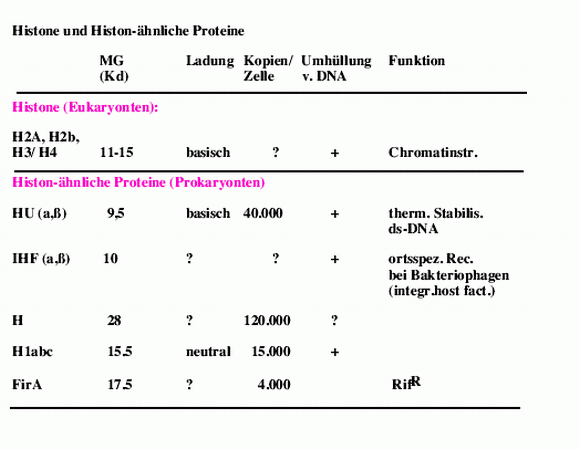
H2A
|
27
|
|
H2B
|
29
|
|
H3
|
30
|
|
H4
|
37
|
|
H1
|
3
|
|
Cytosin
|
14
|
|
Guanin
|
16
|
|
Thymin
|
13
|
|
Adenin
|
7
|
|
|
|
Im Unterschied zu den canonischen Histonen werden die Core-Histon-Varianten nach dem Austausch gegen canonische Histone in bestimmten Phasen einer Mitose vermutlich nur an an einzigen Position modifiziert. Diese Modifikation ist wahrscheinlich mit der Bindung von H1 korreliert. Da der Einsatz dieser Varianten mit großer Wahrscheinlichkeit entweder zellspezifisch erfolgt und/oder auf bestimmte genomische Regionen und Nukleosomengrössen30 beschränkt ist, sind umfangreiche Modifikationen (Acetylierung, Methylierung, Phosphorylierung, Ubiquitinisierung, Ribosylierung u.a.) zum einen nicht erforderlich, zum anderen vermutlich auch kaum in größerem Ausmaß möglich, da zumindest die bis heute identifizierten Histon-Varianten keine tails aufweisen.
|
|
|
Bis heute31 kennt man von H1, H2A, H2B und H3 etwa 20 genetisch kodierte Histon-Varianten. Die Zahl der entdeckten Histon-Varianten steigt aber ständig. Berechnungen, die ich aufgrund von Überlegungen angestellt habe, die ich in Teil II dieser Arbeit (das 2-Zellzyklen-Modell32 ) erläutere, werden (ausser von H4) vermutlich insgesamt 84 verschiedene Histon-Varianten im menschlichen Genom kodiert, die ebenso wie die canonischen Histone posttranslational modifiziert werden. Für diese 84 Histon-Varianten habe ich insgesamt 8433 wahrscheinliche Modifikationen berechnet (jede Histon-Variante wird also wie oben erwähnt - nur an einer einzigen Position modifiziert), sodass sich zusammen mit den oben aufgelisteten Modifikationen der Wert 26034 (176 + 84) ergibt. Der größte Teil der Histon-Varianten und der Histon-Modifikationen ist also noch gar nicht entdeckt.
Wenn man unterstellt, dass die weiter oben genannte Zahl 260 richtig ist und weiter annimmt, dass in der Evolution der menschlichen Zelle jede einzelne dieser Modifikationen für einen bestimmten Entwicklungsschritt während der Phylogenese steht, würde dies gleichzeitig bedeuten, dass 260 Differenzierungsschritte notwendig waren, um aus einer ersten, noch kernlosen Urzelle die menschliche (Ei)-Zelle in ihrer gegenwärtigen Form entstehen zu lassen. (Oder anders ausgedrückt: aus der befruchteten menschlichen Eizelle entstehen im Verlauf der Ontogenese 260 verschiedene Zellen die Zellen der Placenta (mütterlicher und embryonaler/fetaler Anteil ) eingerechnet.)
|
|
Sind Histon-Modifikationen und DNA-Modifikationen gekoppelt?
Die Beobachtung, dass Histon-Modifikationen nicht singulär auftreten, sondern in Kombination mit anderen, regten Strahl und Allis zur Formulierung ihrer Histon-Code-Hypothese an, die allerdings meines Wissens gekoppelte Modifikationen der DNA nicht einschloss im Gegensatz zu der von mit bereits 1991 formulierten Theorie der formatierten DNA, die gekoppelte Modifikationen von DNA und Histonen voraussetzte.
Der von mir 1991 postulierte Histon-DNA-Code sollte also aus einer definierten Zahl von genomischen Schlüsseln bestehen, von denen jeder einzelne für die Kombination einer bestimmten Zahl von spezifischen Modifikationen an der DNA und den Histonen des Oktamers steht und zur Formatierung definierter Abschnitte der DNA mit einer spezifischen Nukleosomengrösse in Phase führt.
Ich werde in dieser Arbeit versuchen, Belege dafür zu liefern, dass die menschliche Zelle eine definierte Zahl35 solcher genomischer Schlüssel nutzt, um durch Modifikation der Nukleotide in definierten Abschnitten der DNA und eine damit gekoppelte Modifikation der Histone in diesen Abschnitten ihr epigenetisches Programm während der Ontogenese zu verwirklichen. Ich werde versuchen zu zeigen, dass die Unterschiede zwischen den verschiedenen Zellen des menschlichen Körpers zu jedem Zeitpunkt der Entwicklung durch unterschiedliche Genomformate36 begründet sind, die wiederum die Ursache unterschiedlicher Expressions-muster sind. Und ich werde zeigen, dass das Genom einer definierten Zelle zu jedem Zeitpunkt der Ontogenese ein mathematisch beschreibbares Genomformat aufweist, das auf der unterschiedlichen Formatierung definierter Abschnitte der DNA beruht.
Gegensätze ziehen sich an.
Grundsätzlich hat die Zelle bei jeder Teilung die Wahl zwischen Differenzierung (wenn ein solches Potential noch vorhanden ist) und Proliferation, das heißt, sie kann sich entweder unter Beibehaltung des Expressionsmusters teilen oder sich unter Veränderung des Expressionsmusters differenzieren. Bei der Proliferation fertigt die Ursprungszelle eine exakte Kopie der Nukleosomen-Konformation ihrer DNA an - bei der Differenzierung ändert sie dieses Muster: definierte Abschnitte der DNA werden so spezifisch mit Nukleosomen besetzt, dass andere Promotoren, andere regulative Sequenzen und andere Poly-A-Signale genutzt werden können (oder müssen).
Wenn, was ja das zentrale Thema dieser universalen Theorie der Genexpression ist, die Anordnung der Nukleotide im Genom nicht nur eine RNA-Chiffre, sondern a priori in Verbindung mit spezifischen chemischen Veränderungen an diesen Nukleotiden und Modifikationen der Histone einen zweiten genomischen Schlüssel darstellt, dann wäre es für die Zelle ein leichtes, den jeweils gewünschten Entwicklungsschritt durch spezifische Positionierung der Nukleosome und eine damit Hand in Hand gehende Modifizierung der Bindungskräfte zwischen den Histonen selbst und zwischen den Histonen und der DNA zu realisieren.
Die Bindungskräfte zwischen der DNA und den Histonen beruhen primär auf Anziehungskräften zwischen positiv oder negativ geladenen Atomen, Molekülen oder Aminosäuren. Die hier angesprochenen Modifikationen verändern diese Kräfte durch gezielte Modifizierung von Aminosäureresten oder von Atomen in DNA-Nukleotiden. Der von mir postulierte Histon-DNA-Code ist deshalb in Wahrheit nicht nur ein quantitativer, sondern auch ein qualitativer Code ein logos, der immanent und von Beginn der Phylogenese an (oder sollte ich sagen, von Beginn der Schöpfung an?) durch die Abfolge der Basen in der DNA (bzw. der RNA) und die zwischen Atomen und Molekülen wirksamen Kräfte festgelegt war und nicht nur die Zahl der Nukleotide definiert, die zusammen mit den Core-Histonen ein Nukleosom bilden und damit den Abstand von Nukleosomenkern zu Nukleosomenkern (die Nukleosomengröße), sondern auch w i e - mit welcher Bindungsenergie - die DNA an das Oktamer und die Histone des Oktamers aneinander gebunden werden und deshalb auch eine Qualität hat.
Denn durch koordinierte Modifikation spezifischer DNA-Nukleotide und von Aminosäureresten der Core-Histone können die Bindungskräfte zwischen der DNA und den Oktameren und jene zwischen den Histonen des Oktamers selbst jeweils so gestaltet werden, dass die Verbindungen abhängig von der Wahl des Entwicklungsschritts entweder fest verdrahtet (um wiederum einen Begriff aus der Elektronik zu verwenden) oder umsteckbar, verschiebbar, löslich sind. Mit anderen Worten, die spezifischen chemischen Modifikationen der DNA und der Core-Histone entscheiden über die Bindungskräfte zwischen den einzelnen Komponenten des Systems und damit auch darüber, ob und wie und unter welchen Bedingungen diese Kräfte zu überwinden sind. Gleiches gilt für die Bindung von H1 an die DNA und das Oktamer.
Ob die Neuorganisation des Genoms durch eine vollständige oder teilweise Aufhebung oder Lockerung der Bindung zwischen den Histonen und der DNA während der Mitose, durch Auflösung der alten und der Generierung neuer Histone oder durch sogenanntes Nukleosomenshifting durch remodeling-Maschinen oder auf eine völlig andere Art bewirkt wird, sei zunächst dahingestellt ( ich werde darauf noch zurückkommen). Das Ergebnis dieser Neuformatierung jedenfalls ist, dass die DNA der betreffenden Zelle nach vollendetem Zellzyklus in definierten Abschnitten ein zellspezifisches Nukleosomen-Muster und damit ein zellspezifisches Genomformat aufweist - ein Muster, das sowohl von spezifischen Modifikationen der DNA-Nukleotide als auch von Modifikationen der Histone des Oktamers abhängt.
.
Die Evolution auch eine Evolution des Histon-DNA-Codes?
|
|
|
|
|
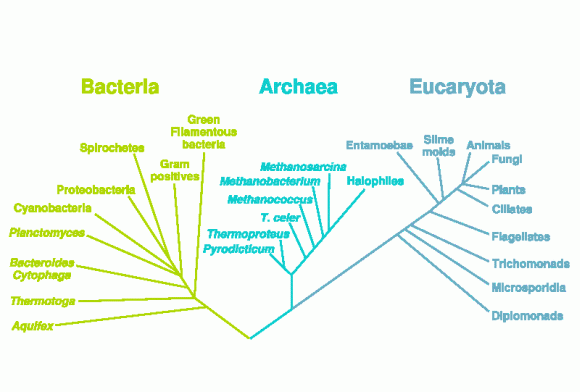
Ob sich zelluläres Leben und damit letztlich auch der menschliche Körper aus einer einzigen Urzelle oder aus unterschiedlichen Lebenskernen entwickelt hat, wird zur Zeit noch diskutiert. Auch der phylogenetische Stammbaum wird durchaus unterschiedlich dargestellt. Das hier gezeigte Entwicklungsschema stammt aus Wikipedia, dass es hier gezeigt wird bedeutet nicht, dass es nach Meinung des Autors die phylogenetisch korrekten Beziehungen darstellt.
Glaubte man bis vor kurzem noch, dass Bakterien keine den Eukarioten entsprechenden Histone aufweisen, so wurde Ende 2005 entdeckt, dass auch Bakterien-Chromosome in Abschnitte eingeteilt sind und eine komplexe Struktur aufweisen, die mit der Funktion der DNA korreliert ist. Histon-ähnliche Proteine und Enzyme, die mit der DNA interagieren, führen zu topologisch unabhängigen Regionen, die einen direkten Einfluss auf die Transkriptionsregulation und die Zellteilung37 haben .
|
|
Geht die Evolution der Genome und damit der Lebewesen also Hand in Hand mit der Evolution des Histon-DNA-Codes?
Dieser Schluß scheint zwingend. Die Formatierung der DNA mit Hilfe eines universellen Codes, der auf der Modifizierung von DNA-Nukleotiden und Histon-Aminosäuren beruht und zur Bildung und spezifischen Positionierung von Nukleosomen oder allgemein zu spezifischen DNA-Protein-Aggregaten führt, scheint ein weitverbreitetes, wenn nicht gar ein allgemeines biologisches Prinzip bei der Organisation der Genome irdischer Lebewesen zu sein. Wenn es richtig ist, dass im Verlauf der Ontogenese die Eckpunkte der Phylogenese reminisziert werden, dann muss sich das Prinzip der formatierten DNA auch in den einfachsten Lebensformen zumindest teilweise noch wiederfinden lassen. Denn was für die Entwicklung vom Einfachen zum Komplexen gilt, muss im Umkehrschluss auch für die Rückführung des Komplexen auf das Einfache gelten. Ich bin mir deshalb sicher, dass der Histon-DNA-Code ein universeller biologischer Code ist, der nicht nur von einem Großteil der Eukarioten, sondern auch von einem Teil der Prokarioten heute noch genutzt wird.
Ich bin mir auch sicher, dass es eine enge Relation gibt zwischen der Komplexität eines Lebewesens bzw. der seines Genoms und der Zahl der Histon- und DNA-Modifikationen und der daraus resultierenden Zahl von genomischen Schlüsseln. In diesem Zusammenhang sei daran erinnert, dass schon lange darüber nachgedacht wird, wodurch die morphologischen Unterschiede zwischen verschiedenen Spezies bewirkt werden, wenn - wie bei Schimpanse und Mensch die Unterschiede in der Struktur der DNA sehr gering sind (die Genome von Mensch und Schimpanse sind zu 98,7 % identisch).
Wie auch Andere schon vor mir ausgeführt haben, ist der Unterschied nicht in der Zahl der Gene begründet, sondern in der Art und Weise ihrer Nutzung. Wissenschaftler des Max-Planck-Instituts für evolutionäre Anthropologie in Leipzig haben die Nutzung von Genen in Körpergeweben verschiedener Spezies untersucht und festgestellt, dass der Grad der Nutzung (messbar an der Menge der produzierten mRNA) der Gene von Leberzellen bei Schimpanse und Mensch kaum differiert, während er in Zellen des Gehirns eklatant ist: Die Gene in menschlichen Gehirnzellen werden offenbar in viel größerem Umfang (etwa um den Faktor 4 stärker) ausgenutzt durch exzessives alternatives Spleißen z.B. als die korrespondierenden Gene der Primaten.38
Offensichtlich hängen die morphologischen Unterschiede zwischen den verschiedenen Lebewesen nicht von der Zahl der Gene ab, sondern vom Grad der Nutzung des vorhandenen Gen-Materials. Nicht die Zahl der Gene ist entscheidend, sondern der Grad der Organisation des Genoms. Und dieser hängt nicht von der absoluten Zahl der Gene ab, sondern von der Zahl der vorhandenen und genutzten genomischen Schlüssel. Vereinfacht ausgedrückt: Evolution beruhte und beruht nicht auf der Evolution der Gene, sondern auf der Evolution der Mechanismen der Genexpression. Deshalb sollte man sich von der (im Augenblick) genannten Zahl menschlicher Gene ca. 30000 bis 45000, je nachdem, wie man das Gen definiert, nicht irritieren lassen. Die Zahl der aus diesen Genen gebildeten mRNAs beträgt nach meinen Hochrechnungen ein Vielfaches, nämlich ca. 245000 (Pawson & Nash, kamen im Jahre 2000 auf ca. 250.000 Proteine), selbst wenn man lediglich jene mRNAs berücksichtigt, die in solche Proteine translatiert werden, die am Metabolismus der Zelle beteiligt sind. Rechnet man die vielen kleinen RNAs dazu, die in irgendeiner Form in die Genexpression involviert sind, kommt man wahrscheinlich auf ca. 350000 Gene das wäre etwa das acht- bis zehnfache des jetzt vermuteten Genpools und dürfte die Wissenschaft vor völlig neue und ungeahnte Herausforderungen stellen.
|
|
9The language of covalent histone modifications; Strahl BD, Allis CD.Nature.
2000 Jan 6;403(6765):41-5.
10Die Literatur-Angaben schwanken zwischen 145 und 147, ich bin bei meinen Berechnungen für die canonischen Histone von einer geraden Zahl, also 146 bp, ausgegangen. Das scheint auch die Grösse zu sein, auf die man sich in den letzten zwei Jahren focussiert.
11Ich definiere die Nukleosomengröße als die Zahl der Basenpaare vom ersten Histon-DNA-Kontakt, der zur Bindung an ein bestimmtes Oktamer führt, bis zum letzten ungebundenen Basenpaar vor dem nächsten Oktamer.
12Hier sind die Cores gemeint
13Czarnota, G.J., Bazett-Jones, D.P., Mendez, E., Allfrey, V.G. and Ottensmeyer,F.P. (1997): High resolution microanalysis and three-dimensional nucleosome structure associated with transcribing chromatin. Micron 28, 419-431.
14Chromatin Immuno-Precipitation
15Dass sich Nukleosomenpositionen dynamisch in einer Zellkultur verändern, haben u.a. Shivaswamy et al. gezeigt: Shivaswamy S, Bhinge A, Zhao Y, Jones S, Hirst M, et al. (2008) Dynamic Remodeling of Individual Nucleosomes Across a Eukaryotic Genome in Response to Transcriptional Perturbation. PLoS Biol 6(3): e65 doi:10.1371/journal.pbio.0060065
16Genome-Scale Identification of Nucleosome Positions in S. cerevisiae;
Guo-Cheng Yuan, Yuen-Jong Liu, Michael F. Dion, Michael D. Slack, Lani F. Wu, Steven J. Altschuler, and Oliver J. Rando (22 July 2005);Science 309 (5734), 626.
17Ich definiere das Nukleosom als Einheit aus Nukleosomencore (146 bp an das Oktamer gebunden) und linker-DNA (einer definierten Zahl von Nukleotiden bis zum nächsten Nukleosomencore).
18Wie wir später noch sehen werden, sind diese Abschnitte Chromatin-Domänen.
19Ich definiere das Nukleosom als Einheit aus Nukleosomencore (146 bp an das Oktamer gebunden) und linker-DNA (einer definierten Zahl von Nukleotiden bis zum nächsten Nukleosomencore).
20Weitere heute bekannte Modifikationen sind Biotinierung, Sumoylierung, Glykosilierung u.a..
21Brownell, J.E., Zhou, J., Ranalli, T., Kobayashi, R., Edmondson, D.G., Roth, S.Y., and Allis, C.D. (1996). Cell, 84,843-851
22Luger et al., 1997. Nature 389:251-260
23http://www.millipore.com/techpublications/tech1/pb1014en00
24http://www.abcam.com/index.html?pageconfig=resource&rid=10583
25Es sollte insgesamt 19 genetisch kodierte humane H1-Varianten geben. Die Begründung werde ich später nachliefern.
26Henikoff,S., Furuyama,T., and Ahmad,K. (2004). Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet. 20, 320-326.
27Siehe: Der Zellzyklus.
28Einen guten Überblick über den Stand des Wissens auf diesem Gebiet geben Rohinton et al. in Histon variants: deviants veröffentlicht in Genes & Dev. 2005 19:295-316.
29Auch canonische Histone genannt
30die NG 204, 208, 212 siehe das Das Nukleosomensystem
31Januar 2009
32Ich nenne es das 2-Zellzyklen-Modell, weil es in meinem Modell einen prämitotischen und einen postmitotischen Zellzyklus gibt. Der postmitotische Zellzyklus endet nicht mit einer weiteren Teilung, sondern mündet in die G0- oder die G1 (Interphase).
33H2A:17; H2B:15; H3:33; H1:19 Varianten
34Die Zahlen 176 und 260 werden später noch eine besondere Bedeutung bekommen.
351114 Schlüssel dies entspricht der Zahl der remodeling-Maschinen, die an der Strukturierung und Organisation des menschlichen Genoms beteiligt sind. Dies wird das Thema inTeil II diese Arbeit sein .
36Die Gesamtheit der verschiedenen Formatierungen von DNA-Abschnitten ergeben in jeder Zelle ein spezifisches Muster: das Genomformat.
37Thanbichler, Wang, Shapiro in J Cell Biochem. 2005 Oct15;96(3):506-21 und Hardy, Cozzarelli in Hardy, Cozzarelli i: Mol Microbiol. 2005 Sep;57(6):1636-52
38Eine Erklärung für dieses Phänomen liefere ich in Teil III dieser Arbeit.
|
|
|