|
|
 |
|
|
|
Wolfgang Marks: Die Formatierte DNA
|
|
|
Teil III.
Der Weg zur Transkriptionsschleife.
In den vorhergehenden Kapiteln habe ich versucht, die Prozesse, Proteine und Hormone zu beschreiben, die an der Formatierung und Strukturierung des Genoms beteiligt sind. Ich hoffe deutlich gemacht zu haben, dass der erste Schritt auf dem Weg zu einem zellspezifischen Primärtranskript und damit letztendlich zu einem zellspezifischen Genprodukt die Formatierung von Chromatin-Domänen mit einer definierten Nukleosomengrösse von einem bestimmten Startpunkt aus im REMAKE der jeweiligen Domäne ist. Diese Formatierung wird durch Formatierungshormone (zu denen auch die bekannten Releasinghormone gehören) oder Silencing-Faktoren initiiert und durch eine große Zahl von remodeling-Maschinen realisiert, die durch die von mir entdeckten REMA-Gene kodiert werden. Diese REMA-Gene sind in jedem Chromosomenband strangweise zu domänenübergreifenden Netzwerken zusammengefasst.
Das bedeutet, dass jeder aktiven Chromatin-Domäne einer gegebenen Zelle und jedem Gen dieser Domäne in jeder Phase der Ontogenese eine Nukleosomengrösse zugewiesen ist, die für den jeweiligen Zelltyp und das Differenzierungsstadium spezifisch ist und sicherstellt, dass auf der einen Seite Promotorsequenzen (TATA-Boxen, TATA-Box-ähnliche Promotoren und Promotoren von REMA- und ALU-Genen164) freigegeben werden, auf der anderen Seite die Transkription an einer spezifischen poly-A-Stelle beendet werden kann.
An der Transkription eines Strukturgens und der Generierung einer zellspezifischen messenger-RNA ist eine Vielzahl von Faktoren beteiligt, von denen viele bekannt, einige wenige und zwar die entscheidenden - allerdings unbekannt sind. Welche Prozesse und Faktoren an der Definition eines Primärtranskripts165 beteiligt sind, auf welchem Wege die Proteine und UsnRNAs aktiviert werden, die bewirken, dass verschiedene Exons zu verschiedenen mRNAs zusammengefügt (gespleisst) werden und wie diese Prozesse mit dem Hormonsystem des menschlichen Körpers verknüpft sind - all dies ist im Wesentlichen nicht gewußt.
In diesem dritten Teil meiner Arbeit werde ich versuchen zu zeigen, welchen Einfluss die von mir in Teil II klassifizierten SMARs und die dort bindenden Proteine auf die Struktur des Chromatins und damit auf die Genexpression haben. Ich will versuchen deutlich zu machen, welche Bedeutung die Formatierung einer Chromatin-Domäne für die Definition einer Transkriptionsschleife, die Bildung eines spezifischen Primärtranskripts und das alternative Spleißen eines solchen Primärtranskripts zu unterschiedlichen messenger-RNAs hat. Ausserdem werde ich zeigen, dass eine andere Klasse von interspergiert repetitiven Elementen (die SINE-Elemente hier vor allem Alu-Sequenzen, aber auch MIR- und andere short repeats), ein den REMA-Genen ähnliches Netzwerk bilden, in welchem wiederum antisense-Promotoren Gene aktivieren, die für Proteine kodieren, die sowohl die Definition und die Transkription einer hnRNA steuern als auch ihr cotranskriptionelles Spleissen zu einer zellspezifischen messenger-RNA.
Und schließlich werde ich ansatzweise versuchen zu erklären, auf welche Weise Hox-Proteine mit der Regulation der Genexpression und mit der Morphogenese des Organismus verknüpft sind.
Das Entpacken eines Chromosoms oder wie aus kondensiertem, inaktivem Chromatin aktives Chromatin wird.
Bevor ich mich den Details der Chromatin-Aktivierung zuwende, möchte ich noch einmal auf die Einteilung der DNA in strukturelle Abschnitte zurückkommen, wie ich sie in Teil II beschrieben habe.
Dass die DNA nicht in gleich große, sondern in unterschiedlich dimensionierte Abschnitte eingeteilt ist, die mit der Kondensation des Chromatins zum Chromosom  und der differentiellen Genexpression verknüpft sind, hatte ich unter anderem in den vorhergehenden Kapiteln zu zeigen versucht. Ich habe die detaillierte Struktur des Chromosomenbandes 15q22.2 aufgezeigt und es mit Hilfe von SMARs, die ich mittels IMPACD® identifiziert habe, in zwölf Transkriptionsgruppen, die Transkriptionsgruppe 11 in drei Chromatin-Domänen und die Chromatin-Domäne 3 dieser Transkriptionsgruppe 11 in vier Subdomänen eingeteilt und postuliert, dass bestimmte Schlüsselproteine und -strukturen diese Einteilung determinieren oder zumindest maßgeblich an ihrer Entstehung beteiligt sind. und der differentiellen Genexpression verknüpft sind, hatte ich unter anderem in den vorhergehenden Kapiteln zu zeigen versucht. Ich habe die detaillierte Struktur des Chromosomenbandes 15q22.2 aufgezeigt und es mit Hilfe von SMARs, die ich mittels IMPACD® identifiziert habe, in zwölf Transkriptionsgruppen, die Transkriptionsgruppe 11 in drei Chromatin-Domänen und die Chromatin-Domäne 3 dieser Transkriptionsgruppe 11 in vier Subdomänen eingeteilt und postuliert, dass bestimmte Schlüsselproteine und -strukturen diese Einteilung determinieren oder zumindest maßgeblich an ihrer Entstehung beteiligt sind.
Um zu verdeutlichen, wie die Zelle die Kern-DNA kondensiert und verpackt, habe ich einige einfache Experimente mit einem Wollfaden gemacht. Der Wollfaden soll das 30nm-Solenoid repräsentieren, also jene Struktur, die wahrscheinlich aus einer verdichteten, etwa 10nm dicken Nukleosomenkette besteht, die in spiraligen Lagen aus je 6 Nukleosomen zu einer ca. 30nm dicken Fiber gewunden ist. Fixiert man ein Ende des besagten Wollfadens auf einer Unterlage und verdreht (verdrillt) das andere Ende mit den Fingern, legt sich der Faden ohne weiteres Zutun wie auf den Fotos zu sehen abhängig von der Länge des Fadens in unterschiedlich grosse Schleifen/loops. Wenn man in bestimmten Bereichen die Struktur des Fadens schwächt zum Beispiel durch Knicken wird sich der Faden beim Verdrillen in Schleifen legen, deren Kreuzungspunkte diesen Stellen entsprechen.
Was zeigt dieses einfache Experiment?
Wenn man in eine wie auch immmer geartete fadenförmige Struktur (sei es nun ein Wollfaden oder die DNA-Helix) eine zusätzliche Windung einbringen will, bedarf es dazu der Fixierung des Objekts an einem Punkt, während am entgegengesetzten Punkt die Kraft ausgeübt wird, mit welcher die Torsion in das Objekt eingebracht werden soll. Die schleifenbildende Wirkung der eingebrachten Kraft ist einmal von der Anzahl der Drehungen, zum anderen aber auch davon abhängig, wie lang das Element ist, auf das diese Drehungen einwirken und von einer eventuell vorhandenen Vorspannung. Ohne Fixpunkte aber, an denen die Kraft ansetzen kann und andere, an denen ihre Wirkung gehemmt wird, würde sich eine eingebrachte Drehung auf die gesamte Länge des Objekts verteilen und wirkungslos bleiben. Eine Schleifen- oder Schlaufenbildung fände nicht statt. Diese Ausführungen gelten sinngemäss auch für den Fall, dass eine Windung aus einer Helix entfernt werden soll oder eine Vorspannung aus einer DNA-Schleife entfernt werden soll.
Dieses einfache Experiment zeigt also, dass die Aktivierung oder die Repression der Genexpression die immer mit der Bildung oder der Auflösung von Schleifen oder loops verbunden ist - in den von mir definierten Domänen und Subdomänen eng verknüpft sein muss mit der Bindung spezifischer Proteine, welche zum einen die Aufgabe haben, Helikasen und Topo-Isomerasen zu rekrutieren, welche die angesprochenen Überwindung(en) erzeugen oder wieder entfernen, zum anderen aber auch die, ihre Weiterverbreitung an definierten Stellen den SMARs - zu hemmen. Auch die Überführung der B-DNA in die Z-DNA-Form, die wahrscheinlich die transkriptionsaktive DNA-Form darstellt, bedarf spezifischer SMARs, an welche die beteiligten Proteine binden können auch um die eingeführten Windungen wieder zu entfernen oder die Konformationsänderung der DNA wieder rückgängig zu machen. So lässt sich die Wirkung der in der Literatur immer wieder apostrophierten Enhancer-Regionen und Isolator-Elemente einfach dadurch erklären, dass so habe ich schon weiter oben argumentiert diese Elemente SMARs mit den weiter oben von mir skizzierten Funktionen darstellen.
Ich hoffe in Teil I und II dieser Arbeit deutlich gemacht zu haben (und ich werde dafür in diesem Abschnitt meiner Arbeit weitere Belege bringen), dass die logische Struktur der DNA zum einen auf einem arithmetischen System von distinkten Nukleosomengrössen beruht, zum anderen durch SMARs bestimmt wird, die man verschiedenen Klassen zuordnen kann und die DNA in Bereiche einteilen, deren Funktionalität von der Bindung spezifischer Proteine an definierte Sequenzen innerhalb dieser SMARs und von der Modifizierung dieser Proteine und der DNA bestimmt wird. Die Bindung solcher modifizierten Proteine innerhalb von SMARs entscheidet darüber, ob die jeweilige SMAR einen reprimierenden oder aktivierenden Einfluss auf den betreffenden DNA-Abschnitt hat.
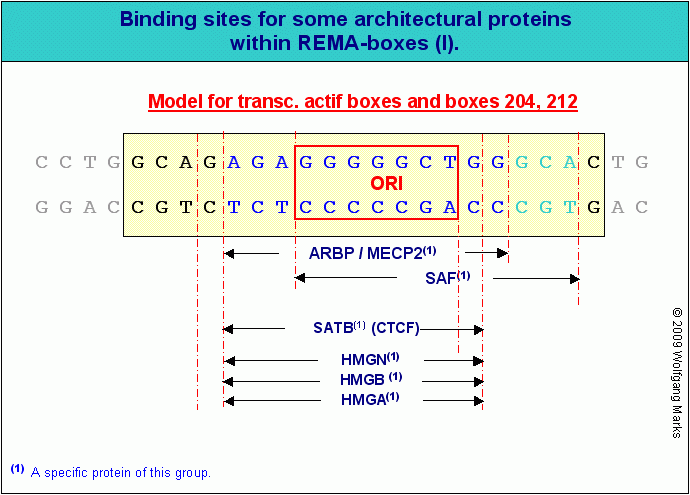
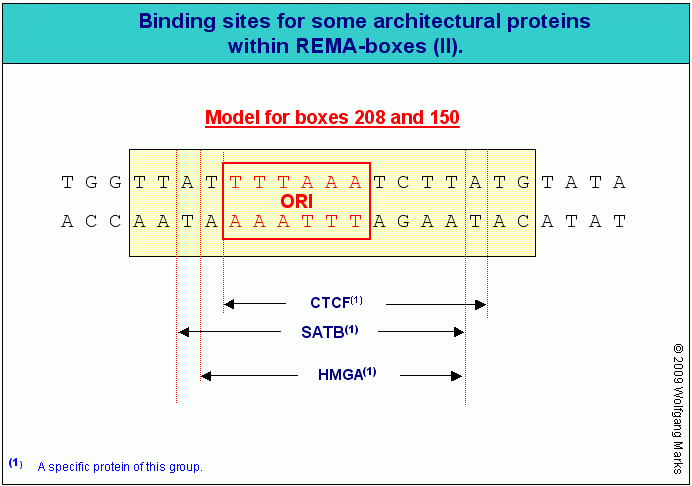
Ich habe postuliert, dass die verschiedenen Protein-Gruppen (HMGA ,HMGB, HMGN, SATB, SAF, CTCF und ARBP/MeCP2 ) zum Teil um die gleichen Bindungsstellen in bestimmten SMARs zum Beispiel in den REMA-Boxen von REMAKEs, aber auch allgemein in den SMARs vor Subdomänen - konkurrieren und durch ihre Bindung und ihre chemische Modifizierung die Funktionalität der korrelierten Chromatin-Region regulieren. Welche Bindestellen in einer bestimmten SMAR genutzt werden, ist demnach wie in Teil II bereits ausgeführt - nicht allein von der topographischen Lage, also vom genomischen Kontext der SMAR abhängig, sondern auch und vor allem davon, in welcher Phase des Zellzyklus ihre spezifische Funktion zum Tragen kommt und welche Proteine in dieser Phase als Bindungspartner zur Verfügung stehen.
So haben zum Beispiel die SMARs/REMAKEs R1 bis R4, die Chromatin-Domänen 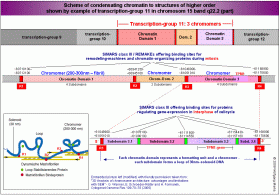 separieren wie wir auf der Abbildung sehen - zum einen eine sehr spezifische Funktion bei der (Neu)Formatierung dieser Domänen mit einer bestimmten aktiven Nukleosomengrösse (ein Prozess, der während der Mitose stattfindet), zum anderen eine ebenso spezifische Aufgabe bei der Steuerung der Expression der Gene der anliegenden Subdomänen in der Arbeitsphase (Interphase) des Zellzyklus. Die SMARs / REMAKEs R1 und R4 haben darüberhinaus Kontrollfunktionen, die damit einhergehen, dass sie nicht nur Domänen- und Subdomänengrenzen markieren, sondern gleichzeitig auch die Grenzregion zwischen der Transkriptionsgruppe 10 und 11 beziehungsweise 11 und 12 darstellen und damit über die genannten Funktionen hinaus auch Aufgaben bei der Festlegung des aktiven DNA-Stranges, beim Imprinting und der Kondensation des aktiven Chromatins zum Chromomer, zum Chromosomenband und zum Chromosom und vice versa bei der Dekondensation des Chromosoms zum aktiven Chromatin haben. separieren wie wir auf der Abbildung sehen - zum einen eine sehr spezifische Funktion bei der (Neu)Formatierung dieser Domänen mit einer bestimmten aktiven Nukleosomengrösse (ein Prozess, der während der Mitose stattfindet), zum anderen eine ebenso spezifische Aufgabe bei der Steuerung der Expression der Gene der anliegenden Subdomänen in der Arbeitsphase (Interphase) des Zellzyklus. Die SMARs / REMAKEs R1 und R4 haben darüberhinaus Kontrollfunktionen, die damit einhergehen, dass sie nicht nur Domänen- und Subdomänengrenzen markieren, sondern gleichzeitig auch die Grenzregion zwischen der Transkriptionsgruppe 10 und 11 beziehungsweise 11 und 12 darstellen und damit über die genannten Funktionen hinaus auch Aufgaben bei der Festlegung des aktiven DNA-Stranges, beim Imprinting und der Kondensation des aktiven Chromatins zum Chromomer, zum Chromosomenband und zum Chromosom und vice versa bei der Dekondensation des Chromosoms zum aktiven Chromatin haben.
Ein und dieselbe SMAR kann also abhängig von der Phase des Zellzyklus und dem dadurch bestimmten Angebot an Bindungspartnern jeweils andere, sehr spezifische Funktionen ausüben. Oder anders ausgedrückt: SMARs der Klasse III die REMAKEs R1 und R4 zum Beispiel - können abhängig von der Phase des Zellzyklus ihre Funktion wechseln. Aus einem mitotischen REMAKE wird in der Interphase eine Subdomänen-SMAR.
Es liegt nahe anzunehmen, dass sowohl am Prozess der Kondensation als auch am Prozess der Dekondensation die gleichen Schlüsselproteine beteiligt sind Proteine, die abhängig von der chemischen Modifikation (Phosphorylierung-Dephosphorylierung, Azetylierung-Deazetylierung u.a.) definierter Aminosäurereste oder der Modifizierung spezifischer DNA-Nukleotide ihre Bindungsaffinität zu bestimmten DNA-Sequenzen und bestimmten Proteinen verändern. Solche Proteine sind mit großer Wahrscheinlichkeit die von mir bereits mehrfach erwähnten Proteine der SAF-, der SATB-, der CTCF, der ARBP/MeCP2-, der HMGA- und der HMGB-Familie, aber auch der HMGN- und NuMA-Gruppe. Zumindest ein Teil dieser Proteine, kann wahrscheinlich das ergibt sich logisch aus allem bis hierher Vorgetragenen - aktivierte hormonelle Faktoren binden (Komplexe aus Rezeptorproteinen und Formatierungshormonen oder aus Rezeptorproteinen und primären Transkriptionshormonen oder aus Rezeptorproteinen und sekundären Transkriptionshormonen). Durch die Bindung solcher Aggregate aus den zuvor erwähnten Proteinen und Hormon-Rezeptorproteinkomplexen an SMARs werden wie wir noch sehen werden Struktur und Organisation des Chromatins im Bereich von Chromomeren/Chromatin-Domänen und von Chromatin-Subdomänen zellspezifisch so verändert, dass die Transkription und das Processing definierter Transkriptionseinheiten in den Subdomänen möglich wird.
Die hierarchische Organisation des Hormonsystems spiegelt sich also in der hierarischen Organisation der Chromatin-Struktur wider - die inhärente Logik der genomischen Datenbank zeigt sich sowohl in der Etablierung von klaren und eindeutigen Ordnungsstrukturen als auch in der Etablierung einer hierarchischen proteinogenen Befehlsstruktur, mit deren Hilfe das komplexe zelluläre Netzwerk über ein System von Schalt- und Schlüsselstellen gesteuert wird.
Das Nukleosomengrössen-System als Basis eines genomweiten
Verpackungs-Systems.
Während der Mitose stellt die Zelle abhängig von ihrem Differenzierungsstatus entweder in einem einzigen mitotischen Zellzyklus (nicht ausdifferenzierte, proliferierende Zelle) die alten Chromatin-Strukturen wieder her oder strukturiert und organisiert das Chromatin während zweier Zellzyklen in bestimmten Abschnitten neu (sich differenzierende Zelle). Ausdifferenzierte Zellen teilen sich nicht mehr166 sie können sich allerdings wieder dedifferenzieren (zum Beispiel beim Wundheilungsprozess) und dann auch wieder proliferative Mitosen durchlaufen.
Wenn die Zelle nach einer Teilung gleich welcher Art wieder in die Arbeitsphase eintreten will, muss sie die Kern-DNA zunächst wieder entpacken. Dieser Prozess ist jedoch mit einiger Sicherheit kein globaler, sondern partiell auf Chromosomen, Chromosomenbänder, Transkriptionsgruppen, Chromatin-Domänen und subdomänen beschränkt, in denen solche Gene liegen, die in der jeweils erreichten Differenzierungsstufe für die spezifische Arbeit der Zelle benötigt werden.
Im aktuellen Stadium nicht benötigte DNA-Abschnitte und endgültig stillgelegte Chromatin-Domänen dagegen werden wahrscheinlich in jeder Zelle als Chromomere aus 30nm-Solenoid-Fibern arretiert. Auch potentiell aktivierbare Chromatin-Domänen liegen vermutlich solange in der Chromomer-Form vor, wie die Gene dieser Domäne nicht benötigt werden. Domäne und Subdomäne werden erst dann entpackt, wenn ein Gen der Domäne oder Subdomäne tatsächlich aktiviert werden soll.
Bevor ein bestimmtes Gen zum Beispiel das TPM1-Gen also transkribiert werden kann, muss das Chromatin des betreffenden Chromosoms, des Chromomers, der Domäne und der betreffenden Subdomäne in einen Zustand versetzt werden, der den Transkriptionsfaktoren den gezielten Zugriff auf die Bereiche der Chromatin-Domäne, der Subdomäne und des Gens erlaubt, die für die Steuerung der Genexpression von der Transkription bis zur reifen messenger-RNA verantwortlich sind.
Dazu werden die nicht stillgelegten Bereiche des Genoms die früher als Euchromatin bezeichneten Abschnitte der DNA - in mehreren Schritten entpackt, deren jeder einer strengen und effizienten Kontrolle durch die Schlüsselmeister des Verpackungssystems den SMARs unterliegt und der mit diesen interagierenden Proteine.
Proteinsynthese erfordert die koordinierte Aktivierung von Genen
über Domänen- und Chromosomengrenzen hinweg.
Viele Polypeptide des menschlichen Organismus werden aus Proteindomänen zusammengefügt, die nicht von einer, sondern von zwei oder drei messenger-RNAs abstammen, die auf unterschiedlichen Genloci und Chromosomen kodiert sind. Dazu kommt, dass fast alle Proteine, welche die Zelle herstellt, zu bestimmten Enzymsystemen (pathways) oder funktionellen Gruppen gehören - ihre Synthese also eine genaue zeitliche (und räumliche) Koordination erfordert. Daher muss es ein umfassendes und vernetztes System geben, das sowohl die kontrollierte Dekondensation des Chromatins als auch die Aktivierung der Gene in den dekondensierten Bereichen und die einzelnen Stufen der Transkription und das Processing der Transkripte koordiniert und kontrolliert.167
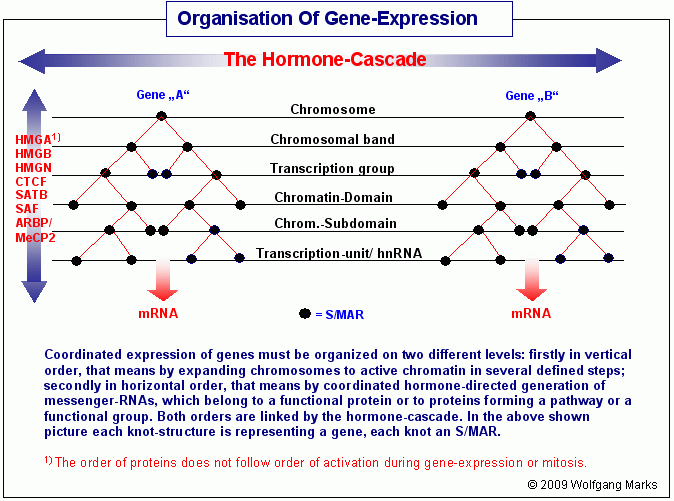
Die logischen Zusammenhänge zwischen den beiden Ebenen der Chromatin-Organisation zeigt die Grafik Horizontale und vertikale Organisation des Chromatins. Die koordinierte Expression von Genen ist demnach auf zwei verschiedenen Ebenen organisiert: einmal in vertikaler (hierarchischer) Richtung durch verschiedene Proteine, welche die Expansion des Chromosoms zum aktiven Chromatin über mehrere Verpackungsstufen steuern, zum anderen in horizontaler (syntaktischer) Richtung durch Remodeling-Maschinen und Transkriptionsfaktoren, die für die koordinierte Synthese von hnRNAs und messenger-RNAs verantwortlich sind. Beide Ebenen sind durch die Hormon-Kaskade aus Formatierungshormonen und primären sowie sekundären Transkriptionshormonen logisch miteinander vernetzt.
Ein Modul dieses Systems haben wir in den vorhergehenden Kapiteln schon kennengelernt: nämlich das Nukleosomengrößen-System, welches was ich hoffe überzeugend dargelegt zu haben mit dem Hormonsystem des menschlichen Organismus aufs Engste verbunden ist. Über das Nukleosomengrössen-System und das mit ihm verknüpfte kaskadierte Hormonsystem lassen sich auf einfachste Weise chromosomenband- und chromosomenübergreifend solche Gene zu Transkriptions- oder Imprinting-Gruppen zusammenfassen, die gemeinsam exprimiert und reguliert werden sollen.
Die Logik sagt uns, dass auch das von mir postulierte zweite Modul des Systems (das in der zeitlichen Abfolge aber an erster Stelle steht) mit dem humanen Hormonsystem korreliert sein muss, denn anders wäre eine koordinierte und kontrollierte Genexpression über die Grenzen von Chromatin-Domänen und Chromosomen-Kompartimenten hinweg wohl kaum realisierbar.
Dieses zweite Modul ist das zuvor schon skizzierte Chromatin-Verpackungs-System der Zelle: eine Art molekularer Packstrasse mit einer Reihe von definierten Weichen (den SMARS), an denen von Fall zu Fall entschieden wird, welche Bereiche des Chromatins für die Arbeit frei gegeben, entpackt werden und welche nicht. Dabei unterscheidet das System ab einer bestimmten Ebene wahrscheinlich variabel zwischen verschiedenen Verpackungszuständen: eine solche Entscheidungs- oder Verzweigungsebene könnte zum einen die 200/300nm-Solenoid-Struktur, das Chromomer, zum anderen die 10nm-Nukleosomenkette mit oder ohne das Histon H1 sein
Konzertierte Aktionen oder wie Architektur-Proteine das Chromatin strukturieren und organisieren.
Von den sieben Stufen168, in denen sich das Entpacken hochkondensierten Chromatins des Chromosoms - vermutlich vollzieht, will ich mich im aktuellen Zusammenhang auf die letzten fünf vom Öffnen der Chromomer-Schleifen bis zur transkriptionsfähigen DNA konzentrieren.
An der Gestaltung dieser Strukturen sind neben den remodeling-Maschinen und Hormon-Rezeptorproteinkomplexen eine Vielzahl von Proteinen und Proteinkomplexen beteiligt, von denen ich die meiner Meinung nach entscheidenden schon mehrfach benannt habe.
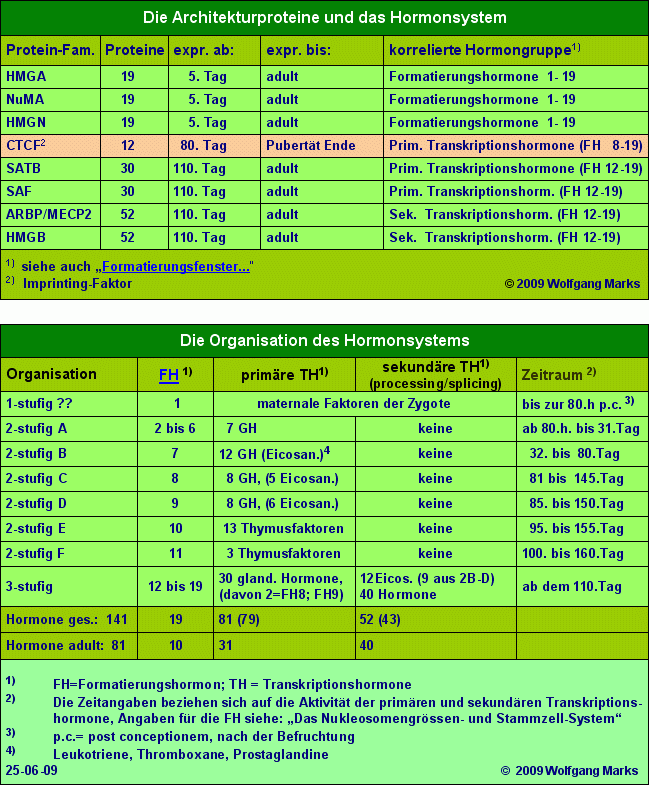
Obwohl ich aus Mangel an zeitlichen und finanziellen Resourcen nicht annähernd alle Details der oben angesprochenen zellulären Packstrasse untersuchen und aufklären konnte, will ich doch versuchen, ein vorläufiges rudimentäres Modell dieses Systems zu etablieren. Es beruht zum einen auf logischen Schlussfolgerungen aus dem bisher Vorgetragenen und eigenen Computer-Analysen, zum andern auf den überaus zahlreichen und fundierten Untersuchungen Anderer zu den Funktionen von NuMA-, HMGN-, HMGA, HMGB-SAF-, SATB-, ARBP/MeCP2 und CTCF-Proteinen. Diese acht Protein-Familien oder gruppen werde ich in Zukunft unter dem Begriff Architektur-Proteine zusammenfassen.
Wenn man sich die Grafik Horizontale und vertikale Organisation des Chromatins vor Augen hält, ist unschwer zu erkennen, dass sich aus der logischen Vernetzung der horizontalen und vertikalen Ebene auch die logische Schlussfolgerung ergibt, dass die Anzahl der Elemente in den einzelnen Gruppen der Architektur-Proteine entweder in direkter numerischer Beziehung zum System der Nukleosomengrössen und/oder in ebenso direkter Beziehung zum kaskadierten Hormonsystem stehen muss.
|
|
|
|
|
Coordinated expression of genes must be organized on two different levels: firstly in vertical order, that means by expanding chromosomes to active chromatin in several defined steps; secondly in horizontal order, that means by coordinated hormone-directed generation of messenger-RNAs, which belong to a functional protein or to proteins forming a pathway or a functional group. Both orders are linked by the hormone-cascade. In the above shown picture each knot-structure is representing a gene, each knot an S/MAR.
|
|
|
Abhängig von der Klasse des SMAR-Elements, an das ein Architektur-Protein bindet und der Phase der Ontogenese, in welcher es bindet, muss es daher in jeder der zuvor erwähnten Proteinfamilien eine definierte Anzahl von Mitgliedern geben. Ein Beispiel: unterstellt, dass jede REMA-Box eine Bindestelle für ein HMGA-Protein aufweist, dann kann es höchstens 23 genetisch kodierte unterschiedliche HMGA-Proteine geben, da es auch nur 23 verschiedene Nukleosomengrössen gibt. Falls die Zahl der HMGA-Proteine geringer ist sollten diese eine entsprechende Anzahl von Bindungsstellen für spezifische Hormone der Hormonkaskade aufweisen. Dies würde alledings nur unter der Voraussetzung gelten, dass HMGA-Proteine oder andere Architekturproteine in allen Phasen der Ontogenese ihre strukturgebende Funktion ausüben.
Auf der anderen Seite sollte von Proteinen, die Subdomänen aktivieren, indem sie an SMARS vor diesen Domänen binden, eine ebenso grosse Anzahl unterschiedlicher genetisch kodierter Varianten existieren, wie es für Subdomänen und für den spezifischen Entwicklungsschritt, mit dem die Gene in Subdomänen generell verknüpft sind, primäre Transkriptionshormone gibt. Da wie ich später noch im Detail zeigen werde - über die Funktionseinheit Subdomäne die Generierung von Primärtranskripten/hnRNAs und das Processing dieser hnRNAs zu messenger-RNAs initiiert wird, müssen an die SMAR, welche die Aktivierung einer solchen Subdomäne steuert, Proteine binden können, die sowohl mit einem übergeordneten Formatierungshormon denn die Subdomäne ist Teil einer Chromatin-Domäne - als auch mit einem von diesem abhängigen primären oder sekundären Transkriptionshormon korreliert sind.
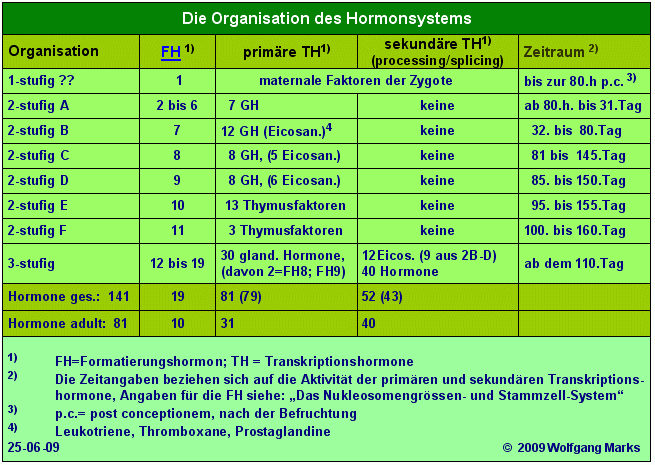
Ein Blick auf die Tabelle Organisation des Hormonsystems aus Teil I zeigt, dass sich die Entwicklung der Hormonkaskade in mehreren Stufen vollzieht, Stufen, denen sich nach dem Ergebnis meiner Computeranalysen bestimmte Hormonklassen und Subklassen zuordnen lassen. Das bedeutet, dass die Proteinfamilien, welche in einer bestimmten Phase der Ontogenese die Funktionalität einer Subdomäne kontrollieren, ebenso viele Mitglieder oder Bindestellen für Hormone haben müssen, wie ich für diese Phase primäre Transkriptionshormone postuliert habe. Diese Feststellung gilt für alle zuvor genannten Proteine, die ich unter den Begriff Architektur-Proteine subsumiert habe.
Jede Architektur-Proteinfamilie muss also eine definierte Zahl von Mitgliedern aufweisen, deren Zahl abhängig von der Funktion des Proteins entweder zu den Nukleosomengrössen (und damit den Formatierungshormonen) in Beziehung steht oder zur Zahl der primären oder sekundären Transkriptionshormone, die mit diesem Entwicklungsschritt und den in dieser Zeit aktiven Formatierungshormonen über die Hormonkaskade verknüpft sind.
|
|
|
|
|
|
Wie ich in Teil II bereits gesagt habe, binden die einzelnen Architekturproteine abhängig vom genomischen Kontext an spezifische SMARs. Dabei konkurrieren oder kooperieren sie von Fall zu Fall mit anderen Architekturproteinen, die wie in der Abbildung modellhaft gezeigt - ebenfalls im Bereich einer SMAR binden können. Einige der genannten Proteine binden nur an REMA-Boxen, die mit einer bestimmten Nukleosomengrösse verknüpft sind, andere binden alternativ oder kumulativ entweder an REMA-Boxen in REMAKEs/SMARs oder in SMARS vor Subdomänen oder in SMARs, welche die Grenzen von REMA-Genen oder ALU-Genen definieren. Auch Modifikationen, wie sie zum Beispiel für die HMG-Proteine beschrieben sind169, haben einen entscheidenden Einfluss auf die Funktionalität der Architektur-Proteine.
Dieses komplexe Geflecht von Beziehungen zwischen Architekturproteinen und SMARs oder die Interaktionen der Architektur-Proteine mit Enzymen oder Remodeling-Maschinen in toto zu analysieren oder zu entschlüsseln, dürfte noch einiges an Aufwand erfordern und ist wahrscheinlich eher durch in vitro-Experimente als durch computational analysis zu leisten.
Die Funktionalität der Architekturproteine lässt sich auch nicht anhand eines einzigen Modells oder einer einzigen Funktionsgrafik erklären: eine umfassende Darstellung der Vorgänge müsste sich auf einzelne Stufen der Ontogenese und der mitotischen Zellzyklen beziehen und würde eine Ausarbeitung erfordern, die der vorliegenden vom Umfang her zumindest vergleichbar wäre. Ich bitte daher um Verständnis, wenn ich hier und heute nur eine auf einen bestimmten Zeitraum und ein bestimmtes Gen fokussierte Darstellung der Zusammenhänge offeriere.
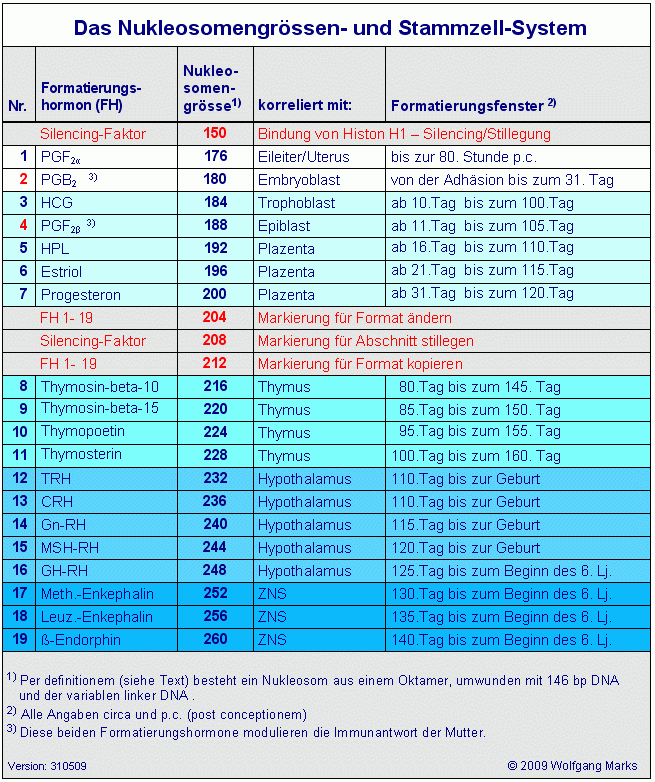
Da für die genannten Architekturproteine keine Daten über ihre Expression in menschlichen Zellen vorliegen, habe ich auf der Basis der über diese Proteine vorliegenden allgemeinen Informationen, auf Grund eigener Berechnungen und logischer Schlussfolgerungen daraus ein System entwickelt, welches die einzelnen Proteine zu bestimmten Phasen der Ontogenese und zu bestimmten Hormonen der Hormonkaskade in Beziehung setzt. Auch bei der Würdigung dieser Tabelle sollte weniger auf einzelne Werte als auf das System geachtet werden, das ich versuche mit diesen Daten deutlich zu machen. Deshalb sollten die Tabellen Die Organisation des Hormonsystems, Das Stammzell-System und die Tabelle Architekturproteine und das Hormonsystem im Zusammenhang gesehen werden.
|
|
|
|
|
164Das vom Autor entdeckte ALU-Gen-System kodiert Transkriptionsfaktoren, die an der Definition eines Primärtranskripts und am Processing desselben beteiligt sind. Das wird später noch ausführlich erläutert.
165Synonyme sind hnRNA (heterogene RNA), prä-mRNA oder pre-mRNA.
166Der Nachschub für ausdifferenzierte Zellen wird von den sogenannten adulten Stammzellen oder von Vorläuferzellen der ausdifferenzierten Zellen geliefert.
167Dieses System kann nicht das Hox-System sein. Wie ich später zeigen werde, regulieren diese Proteine die koordinierte Expression von Genen auf einer anderen Ebene.
168Von der DNA zum Chromosom: I = DNA-Helix (2nm); II = offene Nukleosomenkette (10nm); III = kondensierte 10nm-Nukleosomenkette; IV = Solenoid (30nm); V = Chromomer (200-300nm); VI = Transkriptionsgruppe; VII = Chromosom
169Bergel et al., 2000; Herrera et al., 1999; Lührs et al., 2002; Prymakowska et al., 2001.
|
|
|




{kind=link}
{kind=link}