|
|
 |
|
|
|
Wolfgang Marks: Die Formatierte DNA
|
|
|
Die Formatierte DNA - oder 3,3 Milliarden Basenpaare suchen eine Ordnung.
Was ist ein Gen?
Der Begriff des Gens hat im Laufe der letzten Jahrzehnte eine stetige Bedeutungswandlung erfahren. Vor allem die Entdeckung der gestückelten Gene (früher Mosaikgene genannt - kodierende DNA-Sequenzen, die aus sogenannten Exons und Introns bestehen) und die Feststellung, dass verschiedene Polypeptidketten (Domänen, Precursoren) eines Proteins auf verschiedenen Genloci kodiert sein können, hat die alte Ein Gen ein Protein Definition obsolet werden lassen.
Wie problematisch die heutige Nomenklatur ist, zeigt sich spätestens dann, wenn ein gegebenes Gen in verschiedenen Differenzierungsstufen der gleichen Zelle oder in unterschiedlichen Zellen durch Ansteuerung unterschiedlicher Promotoren und verschiedener poly-A-Signale in unterschiedliche Primärtranskripte und anschließend durch alternatives Spleissen in differente mRNAs und damit Genprodukte übersetzt wird. Das Gen für Tropomyosin alpha (TPM1) zum Beispiel, von dem später noch viel die Rede sein wird, kodiert auf dem plus-Strang des Chromosoms 15 vermutlich ca. 80 verschiedene Polypeptidketten oder Proteine, die in einer großen Zahl von unter- schiedlichen Zellen zu unterschiedlichen Zeiten der Ontogenese gebildet werden. Dass all diese Proteine Varianten der alpha-Polypeptidkette von Tropomyosin darstellen oder Isoformen dieser Kette sind, ist zwar theoretisch möglich, aber unwahrscheinlich. Wahrscheinlicher ist, dass durch TPM1 auch Proteine kodiert werden, die allein oder in Verbindung mit einem anderen Protein eine von Tropomyosin verschiedene Funktion haben. Die (vermutlich) 15 Exons und 14 Introns des Tropomyosin (alpha)-Gens repräsentieren also im Grunde nicht ein Gen, sondern mehrere Gene. Wenn überhaupt, liesse sich ein Gen also nur über eine distinkte messenger-RNA und die durch diese RNA kodierte Aminosäuresequenz eindeutig identifizieren und definieren.
Bei Prokarioten spricht man immer dann von einem polycistronischen Gen, wenn ein prokariotisches mRNA-Transkript die Informationen von mehreren (auf der prokaryo- tischen DNA hintereinanderliegenden) Genen enthält. Die eukariotischen Gene, von denen hier vor allem die Rede sein soll, weisen verschiedene hintereinanderliegende Promotoren und poly-A-Signale auf, deren alternative Nutzung in verschiedenen Zelltypen oder in verschiedenen Differenzierungsstufen der gleichen Zelle zu unterschiedlichen Primärtranskripten führt. Differentielles, alternatives Spleissen dieser Primärtranskripte führt zu differenten messenger-RNAs und damit zu unterschiedlichen Proteinen. Die Prozesse und Mechanismen, die zur Generierung dieser Transkripte und zu ihrer Verarbeitung führen, sind im Großen und Ganzen unbekannt.
Das betreffende gestückelte eukariotische Gen enthält also die Information für mehrere unterschiedliche Proteine. Wenn man sich das vor Augen hält, ist es sicher legitim, bei den sogenannten gestückelten Genen der Eukarioten im Gegensatz zu den monocistronischen Genen (z.B. den Histon-Genen oder den Genen für Regulatorproteine oder bestimmte Hormone) sehr wohl auch von polycistronischen Genen zu sprechen, um damit deutlich zu machen, dass das betreffende Gen für mehrere Primärtranskripte - und demzufolge auch für mehrere messenger-RNAs sprich Genprodukte kodiert. Auch die Verwendung des Begriffs multicistronisch für ein solches gestückeltes Gen, das für mehrere Genprodukte kodiert, wäre denkbar. Da der Begriff polycistronisch aber in der Genetik schon eingeführt ist, werde ich im folgenden immer dann von polycistronischen Genen sprechen, wenn diese aus Exons und Introns bestehen, die zu verschiedenen Primärtranskripten zusammengefasst werden können, die in einem zweiten Schritt zu wiederum unterschiedlichen messenger-RNAs gespleisst werden.
Wie seit langem bekannt und weiter oben bereits erwähnt, können Gene, die für Vorläufer (Domänen, Precursoren) eines bestimmten funktionellen Proteins/Enzyms kodieren, durchaus auch auf verschiedenen Genloci des gleichen Chromosoms oder auf solchen verschiedener Chromosomen liegen. Um gemeinsam reguliert werden zu können, müssen solche Gene sich zum einen auf dem gleichen DNA-Strang befinden (also die gleiche Transkriptionsrichtung aufweisen)3 zum anderen in DNA-Abschnitten organisiert sein, die einer gemeinsamen hormonellen Steuerung unterliegen. Um diese zusammengehörigen DNA-Abschnitte zu kennzeichnen und weil diese Bereiche gemeinsam reguliert und exprimiert werden, verwende ich für sie in dieser Arbeit den Begriff Transkriptionsgruppe.
Datenbank DNA: über drei Milliarden Basenpaare speichern Daten aus über drei Milliarden Jahren zellulärer Entwicklung.
Der Autor, der sich seit mehr als 20 Jahren mit der Auswertung von großen Datenbanken und mit den Programmen beschäftigt, mit deren Hilfe das in überschaubarer Zeit möglich ist, arbeitet seit etwa der gleichen Zeit an einem Computerprogramm, das auf der einen Seite bereits bekannte Verfahren der Gen- und Sequenzanalyse kombiniert (FASTA, BLAST u.a.), auf der anderen Seite aber auch neu entwickelte Verfahren und Algorithmen des Autors in die Berechnungen einführt.
In der mehr als 20-jährigen intensiven Beschäftigung mit dem Aufbau und der Organisation des menschlichen Genoms hat der Autor einen Weg gefunden, die großen Datenmengen des menschlichen Genoms zu analysieren, zu strukturieren und von einem gegebenen Gen ausgehend zusammengehörige Transkriptionseinheiten (also gemeinsam regulierte und exprimierte Gene) zu identifizieren. Durch genomweites screening nach sense und antisense-Promotoren sowohl in sense- als auch in antisense-Richtung der beiden DNA-Stränge ist dem Autor die Entdeckung zweier Netzwerke von sense- und antisense-Promotoren gelungen, deren eines die Synthese von Enzymen steuert, die sich zu sogenannten remodeling-machines zusammenschließen, während das andere die Transkription und Translation von Proteinen aktiviert, die die Definition von Primärtranskripten und deren Spleissen zu zellspezifschen messenger-RNAs regulieren.
Dieses mehrstufige Programm kurz IMPACD® genannt (Integrated Mathematical computational Analysis Of Arithmetic Correlations In DNA) basiert auf der Hypothese, dass die Organisation des menschlichen Genoms einer mathematisch erfassbaren Ordnung unterliegt dass also weder die Nukleotide, noch die Nukleosomen auf der DNA wahl- und regellos, sondern in einer Systematik angeordnet sind, die auf einer mathematisch beschreibbaren Gesetzmässigkeit basiert. DNA ist dieser Auffassung gemäß also mehr als eine RNA-Chiffre, die lediglich abgelesen und durch einen genetischen Code interpretierbar wird: am Anfang des Lebens stand zwar die RNA, aber erst durch die Entdeckung der DNA und die Nutzung der ihr immanenten Möglichkeiten konnte die Entwicklung komplexerer Lebensformen verwirklicht werden.
Die Genialität dieses Schrittes einen redundanten RNA -Tripletcode durch einen quaternären DNA-Code derart zu kodieren, dass sich aus der Anordnung und den chemischen Modifikationen der DNA-Nukleotide in Verbindung mit der Modifikation spezifischer Histon-Aminosäuren wiederum ein Code ergibt - offenbart sich vielleicht nur dem Informatiker, ist aber deshalb nicht weniger bemerkenswert.
|
|
|
|
Auch die Zelle nutzt das Prinzip der Datenkompression.
Eukariotische Zellen speichern ihre Erbinformationen im Zellkern (zum Teil auch in mitochondrialer und anderer extranukleärer DNA).
Im Normalzustand ist die DNA durch definierte Basenpaarungen in Form einer Doppelhelix organisiert. Diese Doppelhelix kann verschiedene Formen annehmen: als A-Form, B-Form und Z-Form bezeichnet.
|
|
|
|
|
|
Wenn DNA nicht gerade abgelesen transkribiert wird liegt sie in der Normal-, der rechtsgewendelten B-Form vor. DNA-Sequenzen, die aktiv transkribiert werden, liegen dagegen wahrscheinlich in der linksgewendelten Z-Form vor.
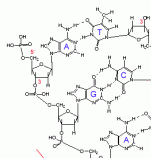
Chemisch gesehen handelt es sich bei der DNA (Abkürzung für englisch: deoxyribonucleic acid) um eine Nukleinsäure, ein langes Polymer aus aneinandergehängten Nukleotiden. Jedes Nukleotid besteht aus einem Phosphat-Rest, einem Zucker und einer von vier organischen Basen mit den Kürzeln A (Adenin), T (Thymin), G (Guanin) und C (Cytosin). Innerhalb von Genen legt die Abfolge der Basen zunächst die Abfolge der Basen im RNA-Transkript fest und falls es sich dabei um eine messenger-RNA handelt - die Abfolge der Aminosäuren des durch das Gen kodierten Proteins.
 Die etwa 3,2 Milliarden Basenpaare des menschlichen Genoms ergäben aneinandergereiht einen ca. 1,09 Meter langen DNA-Faden. Da die somatischen Zellen einen diploiden Chromosomensatz aufweisen, der vor der Teilung noch einmal repliziert, also verdoppelt wird, müssen zeitweilig also ca. 4,4 Meter DNA-Doppelstrang in einem Zellkern mit einem Volumen von ca. 1000 µm3 untergebracht werden. Dies ist nur möglich weil das haploide Genom in 234 hochkondensierten DNA-Protein-Komplexen - den Chromosomen - organisiert ist. Die etwa 3,2 Milliarden Basenpaare des menschlichen Genoms ergäben aneinandergereiht einen ca. 1,09 Meter langen DNA-Faden. Da die somatischen Zellen einen diploiden Chromosomensatz aufweisen, der vor der Teilung noch einmal repliziert, also verdoppelt wird, müssen zeitweilig also ca. 4,4 Meter DNA-Doppelstrang in einem Zellkern mit einem Volumen von ca. 1000 µm3 untergebracht werden. Dies ist nur möglich weil das haploide Genom in 234 hochkondensierten DNA-Protein-Komplexen - den Chromosomen - organisiert ist.
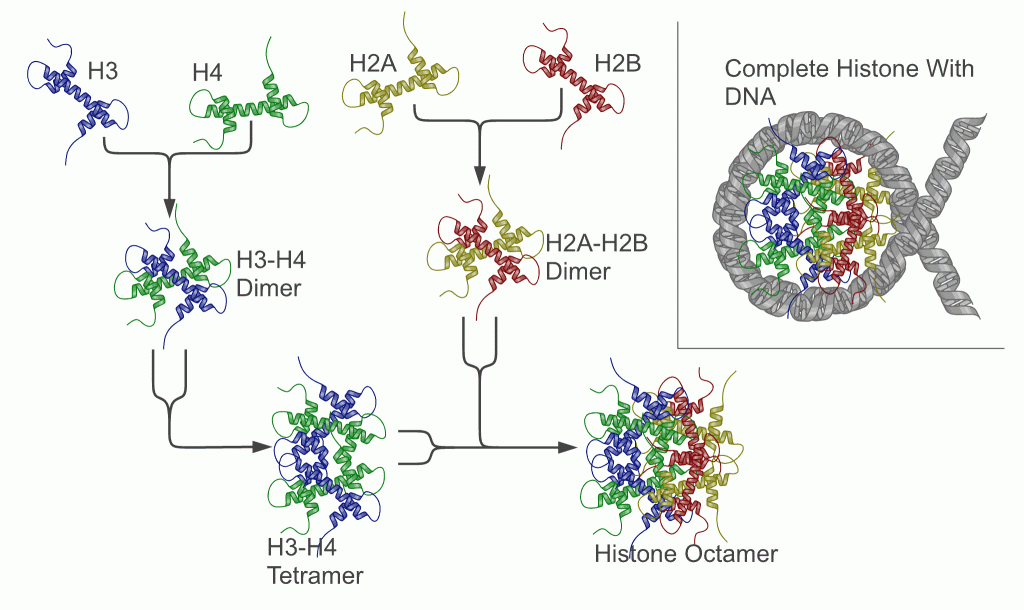
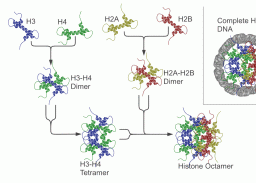
Dass die DNA organisiert ist, weiß man spätestens seit der Endeckung des Nukleosoms und der vier Histone H2A, H2B, H3 und H4, die in aggregierter Form den Nukleo- somenkern (das Histon-Oktamer) bilden (Luger and Richmond, 1998) und insbesondere seit der Entdeckung der Funktion des Histons H1, das eine wichtige Rolle sowohl bei der Verpackung der DNA zum Chromosom, als auch bei der Inaktivierung von DNA-Abschnitten und damit bei der Bildung von (inaktivem) Heterochromatin5 spielt. |
|
|
|
|
|
|
|
|
|
Histon-Proteine schließen sich zu Oktameren zusammen. Um diese herum windet sich die DNA in etwa 1,6 Windungen. Abbildung links aus: aus Wikipedia (Autor: Richard Wheeler (Zephyris) - Abb.: rechts Jeffrey C. Hansen6
|
|
|
|
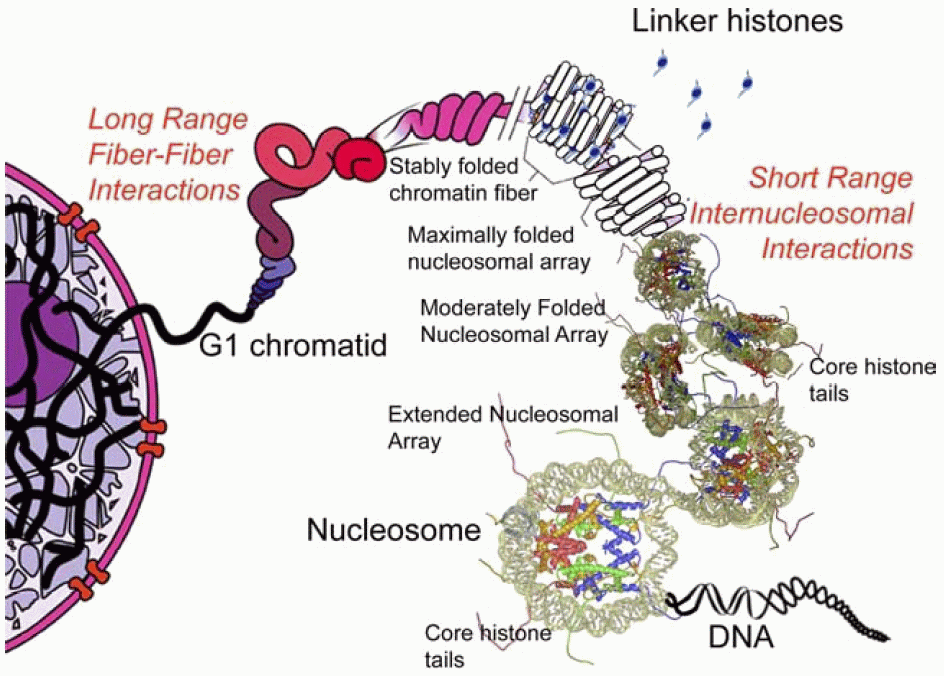
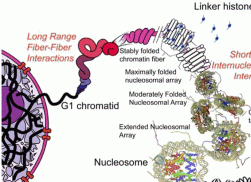
Schon seit längerer Zeit ist in groben Zügen bekannt, wie die eukariotische Zelle ihre Kern-DNA komprimiert, um sie während der Zellteilung korrekt auf die neu gebildeten Zellen verteilen zu können. Dabei spielen die Nukleosomen-Bildung, also die Bindung der DNA an Histon-Oktamere, das Histon H1 in der Abbildung als linker-Histon bezeichnet -, die 10nm-, die 30nm-Faser und höher organisierte Strukturen sowie andere noch weitgehend unbekannte Mechanismen eine Rolle. Wie genau das Chromatin zum Chromosom kondensiert wird, darüber gehen die Meinungen auseinander. Es existieren mehrere, zum Teil sehr ähnliche Modellvorstellungen. Unter diesen scheint mir das Matrix-Fibrillen-Modell von Wanner und Formanek aus den verschiedensten Gründen das am ehesten überzeugende. Dazu später mehr.7
|
|
|
|
|
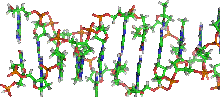
Das Matrix-Fibrillen-Modell von Wanner und Formanek. Abbildung aus: 3D Analysis of chromosome architecture: advantages and limitations with SEM8.
|
|
|
Es wird immer noch diskutiert, ob und wie DNA, die transkribiert wird, in Nukleosomen organisiert ist. Unklar ist auch, wie und warum ein bestimmtes Gen zu einer bestimmten Zeit transkribiert wird und ein anderes nicht. Der Mechanismus der selektiven, differenzierten Genexpression, die Vorgänge, die dazu führen, dass zu definierten Zeiten in definierten Zellen ein bestimmtes Gen exprimiert wird, die epigenetischen Hintergründe der Stammzellbildung und der Genaktivierung sind auch nach oder besser trotz - der Entdeckung der Hox-Gene immer noch ungeklärt.
Dass chemische Modifikationen der Histone bzw. deren tails in Verbindung mit anderen Faktoren zur Aktivierung oder Deaktivierung von Genen führen, gilt heute als weitgehend gesicherte Hypothese. Die verschiedenen Modifikationen der Histone H1, H2A, H2B, H3 und H4 und ihrer Varianten sind zur Zeit Gegenstand des größten wissenschaftlichen Interesses der Histon-Code (das hypothetische System von chemischen Modifikationen der Histone) soll in absehbarer Zeit dazu führen, die Mechanismen der Genaktivierung besser zu verstehen.
|
|
|
3Die Gründe hierfür werde ich später erläutern.
4Der Chromosomensatz einer menschlichen somatischen Zelle in der Arbeitsphase besteht aus 2 mal 23, also 46 Chromosomen. Während der Mitose wird die gesamte DNA repliziert vor der Teilung liegt das Genom also in der Form 4n vor (n=23) bzw. in Form von 46 Zwei-Chromatid-Chromosomen vor.
5Die Unterscheidung von Euchromatin und Heterochromatin als Synonyme für aktives und inaktives Chromatin ist im Grunde überholt. Auch in sogenanntem Heterochromatin gibt es aktive DNA-Abschnitte in Euchromatin inaktive DNA-Regionen.
6Jeffrey C. Hansen, Professor, Colorado State University, Department of Biochemistry and Molecular Biology; Fort Collins, CO 80523-1870; USA
7Siehe Kapitel Die Struktur der DNA
83D Analysis of chromosome architecture: advantages and limitations with SEM; G. Wanner, E. Schroeder-Reiter and H. Formanek, Department of Biology I, Ludwig-Maximilians-Universität München, Munich (Germany) - Cytogenet Genome Res 109:7078 (2005)
|
|
|