|
The aetiology of NCL:
New insights in an old disease.
Identification of new NCL-causing genes and proteins and their implication in different aetiologies and phenotypes of NCL by computational analysis with IMPACD®.
Introduction:
Neuronal ceroid lipofuscinosis (NCL) is an inherited neurodegenerative disorder affecting the central nervous system - but also other tissues and cells - before birth, during infancy or childhood and even in adult age. NCL-diseases share similarities in their pathological and clinical profiles, suggesting a common theme in their disease mechanisms.
NCL is actually thougth to be caused by mutations within 10 different genes - two of them only being assumed to exist and describing a specific phenotype of NCL - being numbered following date of detection from CLN1 to CLN10. Most clinicians use these numbers for characterisation or identification of the disease, besides a classification based on onset and/or country of incidence. Only 4 of these 10 gene-loci one was actually able to associate with a distinct protein or enzyme and also the relation between aetiology of different NCL-variants and function of these proteins or enzymes remained at least partially unclear.
Besides the before mentioned 10 geneloci there are some others, which were brought in connection with onset of NCL like those of the cathepsin-family or the chlorid-ion channel family, but one was until today not able to find a causal linkage between NCL and mutations in these genes.
In this paper, the author of Die formatierte DNA reveals the whole network of NCL-related genes including 8 gene-loci until today unknown for being causally connected with NCL and postulates most of these gene-loci to be linked with known loci on the molecular level, as it was already described for CLN2, CLN3 and CLN530.
Based on computational analysis using IMPACD® (for more information see Die formatierte DNA) the author has detected, that most, if not all NCL-causing genes belong to 4 gene-families, evolutionary being evolved by unequal crossing-over and crossing-over fixing most likely from the common progenitor CLN6.
Intensive computational analysis led to the conclusion, that most NCL-causing genes are coding for an array of proteins - sometimes for a functional isoform, but very often only for single polypeptid chains (in this work called precursors) of specific proteins, which mostly consist of two, three or even four of such polypeptid chains. Using new algorithms and formulas the author was able to identify with a probability of more than 95% besides the just well known four proteins at least eleven more NCL-causing proteins, from which isoforms or precursors are partially coded on known NCL gene loci as well as on gene-loci, actually detected by the author of this site.
In this work, the real causes for the most of NCLs will be described and characterised and the author shows, that similarities between distinct phenotypes of NCL, caused by mutations on different genes, are based on multiple connections between genes and proteins building a large NCL-gene-and-protein network*, in this paper revealed for the very first time.
The common theme in disease mechanisme of NCLs therefore has to be defined by a network of evolutionary closely colligated NCL-causing genes - mostly being linked on the molecular level - which encode a distinct group of proteins, the most pathogenic of them being represented by various isoforms encoded as well on known NCL genes as well as on newly computationally identified by the author.
No single NCL may be caused by a single protein. According to the results of this first comprehensiv computational analysis of the NCL-genes-and-proteins-network the many variants of Neuronal Ceroid Lipofuscinosis from now on have to be characterised as diseases with a multicausal genesis, which have in addition to that very often also a multigenetic base, what will be documented on this side.
Furthermore in this work for the very first time will be offered a convincing theory describing and explaining function and cellular residence of CLN3P, the so called Battenin, the protein believed to be responsible for the most of NCLs, which the author postulates to be a multivalent molecular decyclase, specifically cleaving carbon rings of distinct lipids, in particular those belonging to the terpenes and terpenoids, among which class of compounds many vitamins are counted too.
The author reveals implication of CLN3P in metabolisme of vitamins A/A1, D3, E, K1, K2 and of vitamins of the B-group as well as of carotenes. Function of CLN3P in metabolisme of vitamin D3 and the pathogenic mechanisms leading to a severe longterm D3-hypervitaminosis will be characterised in detail, so giving hope to find a therapy for this and other variants of NCL within a reasonable time.
*naming by the author
 |
|
In Search of Cause of NCL.
Within the last years, gene-prediction has become a wide field in experimental genomic work. Combinating several methods of finding, predicting and analyzing genes has become more and more common to scientist around the world.
The problems ocurring when predicting genes are well known and seemed so far unsoluble. The main reason is, that we know thanks human genom project - very much about the order, in which the nucleotids in DNA, the letters of the genomic language, have been arranged, but not very much about the manner, in which these letters are used to form words or sentences with a meaningful content. One doesnt know very much wether about synchronised regulation and expression of gene-groups nor about organisation of genes in chromatin-domains or -subdomains or how these crucial structural elements are organised in or condensed to chromosomes. Gene expression, in particular when this occurs in form of pre-mRNA or hnRNA-products, which are alternatively spliced to several different mRNAs, is a not at all understood process - predicting of genes or proteins resulting from those pre-mRNAs/hnRNAs was until now extremely difficult and mostly inaccurate.
The author of Die formatierte DNA within 20 years of intensive research - financed by himself - and computational analysis of DNA and thousands of cDNAs of hnRNAs and mRNAs has developed a multistep-program, based on self evolved specific algorithms and formulas, by which he is able to make predictions about genes coupled to transcription groups and to discover relations and linkages between genes and also distinct DNA-sequences one was until today not able to make, respective to detect. IMPACD® enables the author to explore and describe for any given gene complete expression patterns and data as done in Die formatierte DNA for TPM1-gene - that means identification of promotors and terminators (poly-A-signals), definition of transcription windows (transcription loops), hnRNAs/pre-mRNAs and even identification of cell-specific transcription start-sites. In a next step of evolution also definition and prediction of cellspecific mRNAs will be possible by using IMPACD®.
Studying this paper one should remember that human genes are an association of gene-families, which during phylogenesis have been developed and more and more refined by nature - beneath other mechanismes - using unequal crossing-over and crossing-over fixing. Therefore a mutation when being phylogenetic very old - is possibly passed on to a lot of new genes, which have been created by those processes.
As noticed in Introduction I will use in this work the term precursor for characterising a gene-product (a mRNA, translated into a polypeptidchain) which is not representing a fully functional protein, but only a part of such protein - the protein-forming polypeptidchains being coded on more than one gene locus. Approximately 40% of all human proteins being generated during ontogenesis will be constructed from two or three single polypeptidchains, which are coded by separate genes. In this work I will name such premature protein precursor. This remark is valid for all following tables and lists.
What is NCL?
NCL is the short form for a group of diseases with different aetiology, but similar phenotype: the Neuronal Ceroid Lipofuscinoses. With a prevalence of 1:30,000 they are the most abundant inheritable neurodegenerative diseases of childhood and infancy.
The disease is autosomal recessive inherited (at least one form also dominant) and is characterised by progredient mental degeneration, visual failure, movement disorders and epileptic attacks. The onset of different NCL-forms is varying significantly and may lie between early prenatal periods and adulthood. Common to all uptodate studied variants of NCL is supposed to be storage of the so-called ceroid lipofuscin in different cells and tissues of the organism.
Are NCL-causing proteins or enzymes all linked by a common pathway?
Some researchers believe known and until now unknown NCL-causing proteins to be concatenated in a common pathway - or they estimate their individual pathways to belong to linked metabolisms. I will show in this work for he first time, that most of following mentioned NCL-causing or NCL-related proteins are wether genetically linked having common progenitors or are linked on the molecular level underlying common activation and regulation. Furthermore I will show that at least a part of them is belonging to chained or neighbouring pathways and metabolisms.
For many years NCL has been the aim of intensive, but only particular successful research. Until today only for 4 of 10 gene-loci, which are considered to be the cause for the different forms of NCL, one was able to assign a distinct gene-product. These loci are:
|
CLN1
|
Palmitoyl-Protein-Thioesterase 1 (PPT-1)
|
|
CLN2
|
Tri-Peptidyl-Peptidase 1 (TPP-1)
|
|
CLN7
|
MFSD8
|
|
CLN10
|
Cathepsin D (CTSD)
|
|
Most proteins encoded by known CLN-loci are unknown.
Despite of identifying distinct proteins for the known NCL-causing genes the proteins coded by the most of NCL-linked genes have been until today unrevealed.
CLN7 by example is believed to encode the ubiquitous membrane protein MFSD8, which is thought to contain a transporter domain and a so called major facilitator superfamily domain. The substrate(s) of this protein are unknown. Mutations on this gene locus are considered to be the cause for late infantil onset NCL (LINCL).
Gene loci CLN4 and CLN9 are solely defined or postulated by a distinct phenotype of the disease, while the gene products of both genes and also those of CLN3, CLN5, CLN6, CLN8 and CLN9 remained until now unknown.
Defining NCL.
Great efforts were made to get an overview of pathogenesis and symptomatology of different NCLs. The following detailed description, which but leaves some questions unanswered, is based on a classification by gene names and is taken from Neuronal Ceroid Lipofuscinoses, a paper written by Celia H. Chang and published in emedicine from Medscape:
- CLN1 or Santavuori-Haltia type or infantile NCL
- Infantile phenotype
- Retarded head growth
- Hypotonia
- Hyperexcitability
- Cognitive dysfunction
- Visual failure
- Ataxia
- Extrapyramidal movements
- Spasticity
- Myoclonus
- Loss of light perception at age 2 years
- Loss of motor and social skills at age 3 years
- Death between age 6-13 years
- Late infantile phenotype
- Cognitive decline, epilepsy, visual loss at age 1.5-3.5 years
- Resembles CLN2
- Death between age 10-13 years
- Juvenile phenotype
- Visual loss or learning disabilities at age 5-7 years
- Resembles CLN3 except epilepsy later but motor disability earlier
- Adult phenotype
- Starts in third decade
- Psychiatric symptoms with progressive cognitive decline
- Ataxia
- Parkinsonism
- Optic nerve atrophy
- Alive in mid 50s
- CLN2 or Jansky-Bielschowsky type or late infantile NCL
- Late infantile phenotype
- Onset between age 2-4 years
- Epilepsy
- Cognitive decline
- Ataxia
- Myoclonus
- Extrapyramidal symptoms
- Pyramidal symptoms
- Blindness at age 4-6 years
- Death before or in the second decade of life
- Juvenile phenotype
- Onset between age 6-8 years
- Progressive cognitive decline
- Seizures
- Ataxia
- Motor dysfunction
- Variable vision loss
- Survival up to fourth decade possible
- CLN3 or Spielmeyer-Sjögren type or adult NCL
- Classic phenotype
- Progressive visual loss at age 4-7 years, with blindness within 2-10 years
- Speech disturbance
- Cognitive decline
- Epilepsy
- Psychiatric symptoms in 74% of patients, including
social, thought, attention problems, somatic complaints, and aggression
- Parkinsonism
- Myoclonus
- Sleep disturbance
- Pyramidal symptoms
- Cerebellar symptoms
- Extrapyramidal symptoms
- Protracted form - Only visual loss until age 40 years
- CLN4 or Kufs disease or adult NCL - Symptoms usually at age 30 years but can present at age 11 years
- Type A
- Progressive myoclonic epilepsy
- Dementia
- Ataxia
- Pyramidal symptoms
- Extrapyramidal symptoms
- Type B
- Behavior abnormalities
- Dementia
- Motor dysfunction
- Ataxia
- Extrapyramidal symptoms
- Suprabulbar symptoms
- Onset maybe after age 50 years
- CLN5 or Finnish variant late infantile NCL
- Onset at age 4.5-7 years
- Motor clumsiness
- Concentration problems
- Similar to CLN2 but slower course
- Death in second or third decade
- CLN6 or variant late infantile/early juvenile NCL (Lake Cavanagh disease)
- Onset between age 18 months to 8 years
- Visual loss
- Seizures
- Resembles CLN2
- Loss of motor skills between age 4-10 years
- Death in the second or third decade
- CLN7 or Turkish variant late infantile NCL
- CLN8 or Turkish variant late infantile NCL and Northern epilepsy
- Turkish variant late infantile NCL
- Onset at age 3-7.5 years
- Progressive visual loss
- Speech delay
- Seizures
- Intellectual decline
- Myoclonus
- Ataxia
- Northern epilepsy
- Epilepsy at age 5-10 years
- Slight motor dysfunction
- Slowly progressive mental retardation
- May have reduced visual acuity
- May survive to sixth decade
- CLN9 juvenile NCL (CLN3) phenotype
In the above shown list CLN10 is missing, a gene encoding (besides other proteins according to my analysis) cathepsin D (CTSD) - a protein of the large cathepsin family. Its relation to congenital NCL or an NCL with onset in early childhood is in discussion. As we will see later gene CTSD/CLN10 is according to my findings not only coding for CTSD, but for at least five other NCL-related proteins. Cause of CLN10 disease therefore will be discussed further down on this side.
The following table (rtf-document) gives some more informations about the actual state of (gene) research:
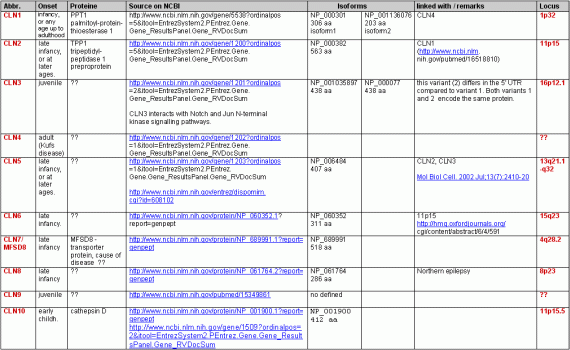 |
Besides the above listed 10 known NCL-causing genes there are some other in discussion to be linked with NCL: these are predominantly genes of the chlorid-ion-channel family and the cathepsin family as well as the SGSH (N-sulfoglucosamine sulfohydrolase) gene, which was recently described to bear two heterozygous mutations in a single patient who was diagnosed with adult onset NCL. But mutations in this gene usually are believed to cause the more severe and unrelated disorder MPSIIIA31.
Single mutations in gene CLCN6 (syn. CIC-6) have been reported in two late onset NCL-patients. Analysis of 75 NCL patients identified CLCN6 amino acid exchanges in two patients but failed to prove a causative role of CLCN6 in that disease32.
Also distinct members of the cathepsin family, comprising up to date 15 different proteins, are besides cathepsin D suggested to be causally linked with NCL. As I will show later, some members of the cathepsin gene-family are part of the NCL-gene-and-protein network too, them encoding proteins - besides a specific cathepsin or a precursor of a protein belonging to that family - which are known or in this paper revealed to be affected by NCL-related mutations.
This statement may be applied - as we will see further down - also for SGSH gene and ARSG-gene, both encoding well known NCL-causing proteins (ARSG) or NCL-related proteins (SGSH) too - but not for genes CLCN6 and CLCN7, which are - based on my calculations - encoding solely proteins belonging to the chlorid-ion-channel protein-family.
Nomenclature of NCL.
For a considerable time classification of Neuronal Ceroid Lipofuscinoses by onset of disease or gene locus turned out to be more and more problematic, because individual aetiologies are difficult to define and to mark off from each other.
The following text is cited from a presentation of Alfried Kohlschütter1, mentor of NCL research, during the 12th International Congress on Neuronal Ceroid Lipofuscinoses (NCL) in june 2009 at Hamburg.2
[..] Using a nomenclature that is illogical and based on the historical order of the recognition of genes, is nevertheless practical, at least in a scientific context.
CLN1 disease, caused by the lack of activity of the lysosomal palmitoyl thioesterase 1, was originally called infantile NCL because its classical manifestation starts in the infantile age. As "infantile" refers to the non-speaking period of human life, this term should not be used for a genetic condition that, depending on the severity of the underlying mutation, can manifest itself at any time between infantile and adult life. The classical form is one of the most dramatic progressive degenerative diseases a pediatrician can be confronted with. It starts in the second half of the first year of life and progresses rapidly with seizures, mental decay, loss of vision, and brain atrophy, the latter recognizable by a falling-off of the head circumference. Some mutations cause manifestation at any age, including adulthood.
CLN2 disease, also called classical late infantile NCL and caused by the lack of activity of the lysosomal tripeptidyl peptidase 1, starts around the third year of life with seizures and a standstill of mental development while the retinopathy frequently is not prominent early in the course and may be missed after progression to more generalized deficits. Patients mostly survive to the age of 10-15 years. Certain mutations lead to later manifestation and a more protracted course.
CLN5 disease, caused by the defect of a partially soluble protein apparently localized in lysosomes, is one of the clinical variants of late infantile NCL that develops symptoms somewhat later than classical CLN2. Regarded as a purely Finnish disease in the past, this type of NCL has recently been observed in the Netherlands, Colombia, Portugal, Italy, Afghanistan, and Pakistan. It should be considered in any exhaustive diagnostic approach to a patient with suspected NCL.
CLN10 disease, caused by the deficiency of the lysosomal enzyme cathepsin D, is the latest on the list of genetically defined NCL disorders and may deserve more attention by the pediatric community. A congenital form is characterized by primary microcephaly, neonatal (possibly already intrauterine) epilepsy, and death in early infancy. Late-onset forms of this NCL may be seen in juveniles and adults. (end of citation)
It is really not easy to mark off the different phenotypes of NCL from each other - NCL in this way has a certain similarity with Multiple Sclerosis (MS) manifesting itself also in many different phenotypes.
Diseases, whose phenotypes or different forms cannot be defined clearly or marked off from each other often have a multicausal or even multigenetic genesis. Therefore we should consider a multifactorial or multigenetic aetiology also for the different variants of NCL, last but not least the different disease patterns being difficult to explain by a single defective or missing protein or by a single genetic defect.
A clear definition and differentiation of various NCLs by symptomatology, time of onset or main area of manifestation is difficult, if not impossible at all.
The NCL-gene-and-protein-network.
Before speaking about the genetic roots of NCL, I may be allowed to cite some sentences from Die Formatierte DNA . I have written there (translated from German):
The terminus gene in the last decades has often got a new definition. Most of all detection of splitted genes (previously named mosaic genes - coding DNA-sequences containing exons and introns) and the finding, that different polypeptidchains (domains, precursors) of a distinct protein may be coded on different gene loci, has made the old definition one gene - one protein obsolete.
The problematic of the current nomenclature is showing off at the latest then, when a given gene in different stages of differentiation of the same cell or in dissimilar cells is transcribed to various primary transcripts and subsequently alternatively spliced to differing mRNAs and afterwards translated to different gene products. This will be done by selection of various promotors and poly-A-signals. The tropomyosin-alpha gene by example (TPM1), which later will be in the focus of this work, is according to my data coding on the plus-strand of chromosome 15 about 80 different transcripts, representing around 50 distinct polypeptides, which are synthesized in many different cells in different stages of ontogenesis.
All these proteins being variants or isoforms of the alpha-chain of tropomyosin may be theoretically possible, but is highly unlikely. More likely TPM1 is also coding for proteins or precursors, which have solely or in connection with another precursor a function different from that of tropomyosin. The (most likely) 15 exons and 14 introns of the tropomyosin-alpha gene therefore are representing effectivly not only one gene, but a certain number of genes. If possible at all, the only way to identify a gene would be to look for a certain messenger-RNA and for the aminoacid-sequence being coded by this mRNA.
[..]
As known for a long time and above mentioned, genes coding for precursors of a distinct functional protein may be positioned on different gene loci of the same chromosome or on those of different chromosomes. To be concertedly regulated such genes must on one hand be located on the same strand (that is to say, they must have the same direction of transcription) - on the other hand they must be organised in DNA-segments underlying common hormonal control and regulation. For characterising such matching DNA-segments, in this work I will use the terminus transcription group. (end of citation from Die formatierte DNA)
Messenger-RNAs of splitted genes are spliced from different exons using the Lego-concept.
All genes until today being considered to be causally for NCL are splitted genes - that means genes containing several exons and introns and being activated, transcribed and spliced following rules and mechanisms I have for the first time described in detail in Die Formatierte DNA.
Splitted genes are functioning (and that is not new) following the Lego-concept: depending from stage of differentiation, from cellular context and from hormonal and other signals the cell is assembling various exons of a (virtual) primary transcript (hnRNA, pre-mRNA) to different messenger-RNAs. After translation of a messenger-RNA the resulting protein case by case will be chemically modified mostly on different positions, leading to protein-products with possibly different functions. Very often those posttranslational modifications will be used as a code for trafficking purposes - in this way marking a specific protein to be transported to a specific cell organelle or a distinct working place.
Also Enzymes or Proteins are assembled following the Lego-concept.
In a similar way as the cell is combinating different exons to different messenger-RNAs the cell assembles different protein-precursors represented by different polypeptidchains being coded on different gene loci - by this manner constructing new proteins or enzymes. In this context it may be allowed to remember, that the number of proteins being coded by (estimated) 40,000 metabolic genes is much more bigger than this genepool. Considering only 60% of metabolic genes being splitted genes this means, that any splitted gene on average is coding for more than 9 messenger-RNAs, if we set the number of proteins involved in metabolism of human cells to 245,000, as I have done in Die formatierte DNA, or to 250,000, as Pawson & Nash4 have done in year 2000. Including also posttranslational modifications of these proteins in our calculation we will count circa 400,000 different proteins produced in all together 260 human cells being evolved during ontogenesis. Working this out, around 15 different proteins (including posttranslational modifications) should be apportioned on average to each splitted gene. (Setting 50,000 proteins being coded by non-splitted genes the calculation is as following: 60% of 40,000 genes = 24,000 genes; 400,000 proteins minus 50,000 = 350,000./. 24,000 = 14,58). Comparing this data with identified or characterised gene products, it becomes obvious, that a lot of work in detecting proteins coded by human genes remains to be done...
The definition of a certain primary transcript (syn.: hnRNA, pre-mRNA) and splicing of such transcript to different cellspecific messenger-RNAs ist primarily regulated - as being described in detail in Die Formatierte DNA - by a cascaded system of hormonal signals and more than this, also the pooling of chromosomal-overarching transcription groups and coordinated, synchronized activation of those groups is controlled by a hormone cascade. Therefore it may not be unlikely, that one chain of two or even three chains belonging to a distinct protein or enzyme will be defective, while the other chain(s) coded on different location(s) are free of faults.
Most proteins or enzymes being involved in cell metabolism exist in various, mainly cell- or tissue-specific isoforms, which also depend from phase of development. Protein disulfide isomerase by example is known to exist in 17 isoforms until today - and existence of more of those isoforms is presumed.3
Why are different isoforms of a certain protein or enzyme required?
During ontogenesis in human organism nearly all cells are undergoing a steady development - depending from internal and external signals they arise from undifferentiated or less differentiated forms during morulagenesis or embryogenesis to highdifferentiated and specialized cells of the adult organism. Parallel to this development, metabolism and catalytic pathways of those cells will be steadily modified: many cycles or chaines of reaction (pathways), in which enzymes and/or proteins are collaborating in a well coordinated and controlled manner, are differently organised and structurised in different stages of ontogenesis and therefore require use of different isoforms or different modified isoforms of a given protein catalyzing a defined reaction in such pathway. CLN3 protein (CLN3P) - the so called Battenin, thought to cause juvenile NCL - may serve as a good example for that behaviour.
Also NCL-causing proteins are existing in many isoforms.
As above mentioned, all known and also the here newly revealed NCL-related genes are splitted genes: they consist of a more or less great number of exons and introns. Splitted genes are often coding for a precursor - a single polypeptidchain - of a protein, whose functional version is build from two or three of those precursors - so not coding a per se functional protein.
Identifying those proteins being assembled from one or more different polypeptidchains generated by different genes is not only very difficult for molecular biologists, but also for computer scientist and geneticists. This may be one of the reasons the enormous number of multiple linked networks of those Lego proteins existing in human organism have not been detected by far until now.
Primary and secundary genetic defects.
Not any genetic defect is equal: mutations may affect regulatory DNA-sequences (secundary defects) as well as the nucleotide sequence of the gene itself (primary defects). Therefore it makes a difference the mutation affecting a promotor region, a transcription start site, an ALU-gene, an SMAR or the DNA-sequence inside of an exon-intron territory. Because one did not know much about mechanisms of gene expression before publication of Die formatierte DNA, one also did not know much about influences of mutations on gene expression, when such mutations were located in those extragenic regulatory regions like REMAKEs or SMARs - sequences, I have characterised and described in detail in my cited work for the very first time.
Therefore we should consider, that NCL causing mutations may also exist in regions I have previously defined in Die formatierte DNA as ALUgenes or REMAgenes and in regions like SMARs or REMAKEs - segments crucial for setup and controlling of human gene expression.
Depending from type of genetic defect - from being a primary or secundary defect, depending from location of the mutation in extragenic or intragenic sequences, in an intron or in an exon, a genetic defect may have very different effects on disease patterns or its progression: this is spanning from no failures over little failures to letal forms of the disease. Function of a protein may be more or less affected - and total loss of function of more than one distinct protein caused by a single mutation in a specific gene may also be possible. The latter scenario may take place by example when a defective polypeptid chain (precursor) is being used for composition of various other proteins as it most likely is the case with NCL.
It is also possible - and this phenomen may play a role in aetiology of NCL too - that from various products of a given gene one is hit by a defect, while another product of the same gene is not affected. This is without any doubt the fact in regard to CLN3, where the well known 1 kb deletion does indeed affect the identified CLN3P 438aa protein (and possibly other proteins - see tables below and data tables), but possibly not those gene-products being build from exons, which are not hit by the 1 kb deletion. This statement is valid in a transferred sense for all NCL-causing or NCL-related genes.
Mutations up to date detected for known NCL-genes are listed by the NCL mutation database of the University College London (http://www.ucl.ac.uk/ncl/mutation.shtml), which is in steady actualisation.
|
Computational analysis of phylogenetic and molecular relationships between those genes newly detected by the author and known NCL-related genes reveals a large-scale genetic network of NCL-causing genes and proteins.
As mentioned before, within more than 20 years I have developed a multistep program combinating several known procedures for analysing sequences and proteins like BLAST, FASTA and others with algorithms based upon the hypothesis, that the human genome exhibits inherent logical structures and a syntax - in principle therefore functioning like a database. Using IMPACD® I am able not only to identify gene-families emerged from unequal crossing over and crossing over fixing, but also genes belonging to distinct transcription groups, them being therefore regulated and expressed in an coordinated manner. Based on a self-developed time-oriented pedigree of human cells (unpublished until today) IMPACD® also enables me to define potential expression data for distinct proteins encoded by those genes, which have high probability.
Identification of NCL-causing genes until now unknown with IMPACD®.
According to my data based on computational analysis the below listed 24 NCL-related genes have arised from the common progenitor CLN6 resulting in four big trees of genetic evolution.
As we may see in the following diagram, there are in total 4 lines of genetic development, which arised from unequal crossing over events and crossing over fixing.
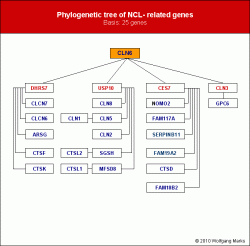 Many of mutations having accumulated in CLN6, CLN3, USP10, CES7, DHRS7 and other genes within millions of years were relayed by unequal crossing over events to their duplicates. Because most of NCL-diseases were autosomal recessive inherited such genetic defects could survive, even most of these defects being letal prenatal, during childhood or during adolescence. Many of mutations having accumulated in CLN6, CLN3, USP10, CES7, DHRS7 and other genes within millions of years were relayed by unequal crossing over events to their duplicates. Because most of NCL-diseases were autosomal recessive inherited such genetic defects could survive, even most of these defects being letal prenatal, during childhood or during adolescence.
Therefore NCL-causing genetic defects very often should emerge coupled. Roughly calculated, around 70% of the NCL-diseases should have a multicausal genetic basis. This refers as well to the coupling of genetic defects on evolutionary closely linked genes like CLN3/GPC6 or CES7/NOMO2 as to the (non)activation of different defective proteins in different cells with also different functions coded completely or in part (precursor) on the same gene. The CLN3-gene by example is according to my evaluations besides coding precursors for various NCL-causing proteins like acetoin racemase, multivalent terpene and terpenoid decyclase, cathepsin D or tripeptidyl-peptidase also coding functional isoforms of PPT, retinol isomerase, CTSL2, CLCN7 and four different isoforms of multivalent terpene and terpenoid decyclase (for full information: see table below).
The effects of up-to-date detected mutations therefore not being restricted to a single protein, but following the Lego-concept being spread over two or even three or more proteins, whose precursors or isoforms are coded on the same or different genes, symptoms being generated by individual defective proteins interfere. This may be one of the main reasons for the indifferent symptomatology of most lipofuscinoses. These context will be discussed later by example of genes CLN3 and USP10.
A given mutation in a distinct gene must not affect all proteins generated from this gene during ontogenesis.
Regarding the above shown pedigrees of NCL genes and regarding the multiple relationships and cross-connections between these genes shown later on this site it may be difficult to distinguish between NCL-related and NCL-causing genes on one side and distinct NCL-causing isoforms of a defined protein on the other side.
Depending from expression patterns, one isoform of a certain protein may be unusable due to a mutation, while another one may be fully functional. Not any given mutation in a given gene must affect all proteins being generated from this specific gene during ontogenesis.
Because NCL is a disease predominantly ocurring during all prenatal and postnatal stages of ontogenesis we should consider, that in one step of cell development a certain isoform of a given protein may work properly, while in next step of cell development the following isoform of the same protein may be defective. In this case it will be important to know expression patterns when searching for a distinct defective protein.
The following list is divided into two parts:
- NCL-related genes and genes with NCL-related mutations
postulated by the author.
- Known NCL-causing genes with known mutations too.
In the following two tables I differentiate between genes I postulate to bear NCL-related (causing) mutation(s) and genes I postulate only to be linked with such genes on the molecular level - they themselves most likely not bearing any NCL related mutation. The former I have labeled NCL-related mutation(s), the latter NCL-related. In regard of the above mentioned correlations between a given mutation and a specific isoform of a certain protein NCL related mutation means, that the specific gene resp. one or more of the proteins encoded by this gene are involved in onset and progression of one or even various variants of NCL.
All genes on the following table are postulated by the author to belong to the NCL-gene-and-protein network. Nine of these genes (marked red) must be involved in onset and/or progression of distinct variants of NCL, as it will be discussed in detail further down on this side.
|
GPC6
|
13q32.1
|
NCL-related mutation(s)1)
|
|
DHRS7
|
14q23.1
|
NCL-related mutation(s)
|
|
NOMO2
|
16p13.12
|
NCL-related mutation(s)
|
|
CES7/CES5A
|
16q12.2
|
NCL-related mutation(s)
|
|
USP10
|
16q24.1
|
NCL-related mutation(s)
|
|
CLCN6
|
1p36
|
NCL-related mutation(s)2)
|
|
CLCN7
|
16p13
|
NCL-related mutation(s)2)
|
|
CTSK
|
1q21
|
NCL-related mutation(s)2)
|
|
CTSF
|
11q13
|
NCL-related mutation(s)2)
|
|
ARSG
|
17q24.2
|
NCL-related
|
|
SGSH
|
17q25.3
|
NCL-related3)
|
|
FAM18B2
|
17p12
|
NCL-related
|
|
FAM19A2
|
12q14.1
|
NCL-related
|
|
CTSL1
|
9q21-q22
|
NCL-related
|
|
CTSL2
|
9q22.2
|
NCL-related
|
|
SERPINB11
|
18q21.33
|
NCL-related
|
|
FAM117A
|
17q21.33
|
NCL-related
|
|
1) Those defects may also be mutation(s) in SMARs building 5-border of respective subdomain and also in SMAR (REMAKE) of superior domain, comprising such subdomain. For detailed information about organisation of domains in human genome see here.
2) Though CLCN6 (CIC-6) is bearing single mutations a causative role for onset or progression of NCL could not be proved32. Because CLCN6 is according to my calculations encoding 11 different transcripts for proteins of the chlorid-ion-channel family (including 8 isoforms of CLCN6 - see here) the role of CLCN6 in development of this disease should be kept in view.
I found CLCN7 to encode transcripts for a great number of proteins belonging to the chlorid-ion channel family - any mutation in this gene should have severe consequences for establishing those channels in lysosomal membranes or other membranes of distinct cells.
Cathepsins in general are in discussion to be connected with different phenotypes of NCL. Several papers deal with this theme.36 Following my own findings and calculations (see analysis of genes further down) I postulate different cathepsins to be involved in onset and/or progression of various NCLs. Defects/mutations in these two genes may occur coupled according to their emergence from the common progenitor DHRS7.
3) I postulate SGSH to belong to the NCL genes network, but not to be causally involved in onset or progression of this disease, even there were found mutations in NCL-patients31. But in general this distinction is depending from a strict definition of NCL.
|
|
These are known NCL- genes, all bearing different mutations:
|
CLN1/PPT1
|
1p32
|
NCL-related mutation(s)
|
|
CLN2/TPP1
|
14q23.1
|
NCL-related mutation(s)
|
|
CLN3
|
16p12.1
|
NCL-related mutation(s)
|
|
CLN5
|
13q21.1 - q32
|
NCL-related mutation(s)
|
|
CLN6
|
15q23
|
NCL-related mutation(s)
|
|
CLN7/MFSD8
|
4q28.2
|
NCL-related mutation(s)
|
|
CLN8
|
8p23
|
NCL-related mutation(s)
|
|
CLN10/CTSD
|
11p15.5
|
NCL-related mutation(s)
|
|
Most proteins encoded by the above listed genes are linked on the molecular level. They form a large protein-network.
While nowadays it seems to be quite easy to analyse a distinct gene of an NCL-patient for being hit by a mutation or not, it is - for reasons I have discussed before - not as easy to decide, which specific protein(s) generated from this specific gene may be affected during ontogenesis by a distinct mutation or not.
Interactions between different geneloci on the molecular level have been described already in 2002 ((Mol Biol Cell. 2002 Jul;13(7):2410-20). The authors refer to polypeptides being coded by genes CLN2, CLN3 and CLN5 that are interacting in various manners. CLN5 is supposed to generate 4 different precursors - based on their evaluations the authors conclude CLN5 most likely being an membran-bound lysosomal enzyme. CLN1 is beyond that thought not only to be responsible for the CLN1-variant of the disease, but also for CLN4 - a NCL of the adult age (Kufs disease). Furthermore, expression of CLN2 is thought to be correlated with CLN1.5
During the 12th International Congress on Neuronal Ceroid Lipofuscinoses (NCL) in june 2009, Hamburg, interactions finally were reported also between CLN5, CLN1 and CLN8.6
My own data, based on computational analysis with IMPACD® and presented further down on this side, show these interactions not only for the above mentioned five genes, but for all 25 genes presented to belong to the NCL-Network on this side.
My data and evaluations show besides the well known proteins TPP, PPT, MFSD8 and Cathepsin D a great number of until now unknown NCL-related or NCL-causing proteins, most of them being represented by multiple isoforms coded on one or more of the above listed 25 geneloci. My data also clearly demonstrate, that also from the well known proteins PPT, TPP, CTSD and MFSD8 are existing various isoforms, so building - together with the actually by the author detected proteins - not only a genetic but, at least including most of them, also a metabolic or catalytic network in distinct cells and in this way causally participating in onset and or progression of different variants of NCL.
The following table counts 28 different proteins, most of them being linked on the molecular level by precursors coded on one or more of the above listed genes. Though some of these proteins - on the first view - may appear to be implausible or even odd, I am strongly convinced to be on right side even with such exotic, but known proteins like beta-L-arabinosidase, acetoin racemase, deoxyribonuclease or the until now unknown and initially by the author postulated and characterised multivalent terpene and terpenoid decyclase, a specific isoform of the latter being most likely identic with CLN3P (438aa), a protein whose function one was trying to detect for a long time. This items will be discussed in detail later on this site.
The relations between the above mentioned genes and the below listed proteins will be shown further down in an overview of all NCL-related gene loci and their calculated gene products, as far I was able to identify them by computational analysis until now.
Even if a gene itself shows no mutation, it may generate a defective protein - or not a protein at all - due to a linkage with a mutated gene.
A gene discussed on this side not being hit by a mutation does not mean this gene not being involved in onset or progression of any NCL. As I will show later on this side, from most proteins listed in the NCL-Protein-Network table below exist various isoforms, from which very often one precursor is encoded on a gene bearing no NCL-related mutation, while the other precursor might be encoded on a gene with such defect.
The gene SERPINB11 itself by example is - according to my calculations - not hit by a NCL-relevant mutation, but is however participating in causing NCL by coding isoform 14 of multivalent terpene and terpenoid decyclase, which should be build from two polypeptidchains, one coded on gene SERPINB11 and one on USP10. (For this reason I have characterised SERPINB11 as NCL-related only.) Although SERPINB11 itself bearing no NCL-related mutation, this isoform 14 of MTTDC - which will be activated beginning with end of year 5 - will be missing totally in patients with a distinct mutation in gene USP10. For better understanding of those coherencies You may have a look at the data table for MTTDC.
Proteins (including their isoforms) in the following list marked red are believed by the author to be involved in onset and/or progression of one or more variants of NCL. Depending from definition and from including or not also milder or very rare forms of this disease 16 different proteins with their various isoforms according to my calculations and evaluations should therefore be involved in aetiology of - estimated 20 - different genetic variants and 13 different phenotypes34 of NCL worldwide.
|
The NCL-Protein-Network1)
|
|
Protein family
|
Abbrev.
|
EC Number
|
|
acetoin racemase
|
AR
|
5.1.2.4
|
|
arylsulfatase G
|
ARSG
|
3.1.6.1
|
|
beta-L-arabinosidase
|
BLA
|
3.2.1.88
|
|
carboxylesterase 7
|
CES7
|
3.1.1.1
|
|
cathepsin D
|
CTSD
|
3.4.23.5
|
|
cathepsin F
|
CTSF
|
3.4.22.41
|
|
cathepsin K
|
CTSK
|
3.4.22.38
|
|
cathepsin L1
|
CTSL1
|
3.4.22.15
|
|
cathepsin L2
|
CTSL2
|
3.4.22.43
|
|
chlorid-ion channel 6
|
CLCN6
|
none
|
|
chlorid-ion channel 7
|
CLCN7
|
none
|
|
dehydrogenase/reductase member 7
|
DHRS7
|
none
|
|
deoxyribonuclease type I, site-specific
|
none
|
3.1.21.3
|
|
CC-preferring endodeoxyribonuclease
|
none
|
3.1.21.6
|
|
family with sequence similarity 18, m. B2
|
FAM18B2
|
none
|
|
family with sequence similarity 19, m. A2
|
FAM19A2
|
none
|
|
family with sequence similarity 117, m. A
|
FAM117A
|
none
|
|
glypican-6 precursor
|
GPC6
|
none
|
|
major-facilitator-superfamily-domain containing 8
|
MFSD8
|
none
|
|
multivalent terpene and terpenoid decyclase2)
|
MTTDC
|
5.5.1.??
|
|
NODAL modulator 2
|
NOMO2
|
none
|
|
N-sulfoglucosamine sulfohydrolase
|
SGSH
|
3.10.1.1
|
|
palmitoyl-protein-thioesterase (-hydrolase)
|
PPT
|
3.1.2.22
|
|
protein disulfide-isomerase
|
PDI
|
5.3.4.1
|
|
retinol-isomerase
|
RI
|
5.2.1.7
|
|
serpin peptidase inhibitor, clade B, m. 11
|
SERPIN B11
|
none
|
|
tripeptidyl-peptidase
|
TPP
|
3.4.14.9
|
|
Ubiquitin specific peptidase 10
|
USP10
|
3.1.2.15
|
|
1) Proteinfamilies - most of them linked on the molecular level by their coding
genes, each comprising various isoforms, sorted by alphabet.
2) Naming by author -former a-pinene-oxide decyclase. From now on this site
this enzyme will be characterised as multivalent terpene and terpenoid decyclase
(MTTDC), for further explanations see separate chapter for CLN3 further down on this
side.
|
|
The above listed proteins - most of them for the first time brought in connection with NCL - according to my evaluations must besides the usual suspects TPP, PPT, CTSD and MFSD8 be more or less involved in the aetiology of until now familiar and eventually still unknown or unverified very rare NCL-variants, because all genes coding for those proteins show - as I will demonstrate below - multiple cross-connections on the molecular level, some of those connections (CLN1, CLN2, CLN3, CLN5, CLN8) just being revealed by other authors.
As I have discussed before, there are existing many various isoforms of all NCL-related proteins. For some proteins I have tried to define isoforms based on logical rules and coherencies I have described in Die formatierte DNA. I have also tried to calculate expression patterns (see data tables), which may give an explanation for some until now inexplicable and contradictory phenomens occurring in some variants of NCL. Because calculation of those expression data is a very time-robbing and stressing issue for the moment I have focussed my activities on AR, CTSD, MTTDC, PPT, RI and TPP, proteins (including their isoforms) which should according to my evaluations be responsible for around 80% of all NCL diseases worldwide.
According to my updated calculations based on IMPACD® the above presented list of 28 NCL-related or NCL-causing proteins must be expanded to approx. 50 proteins, if we include all their isoforms, them all together building a large overarching protein network during ontogenesis.
|
Overview of the NCL-Gene-and-Protein-Network, linked geneloci and NCL-causing proteins or precursors coded on these loci.
Preliminary remarks:
Since my last efforts to analyse NCL-geneloci in detail much time has gone. In this time the universal database, IMPACD is working with, has undergone comprehensive extension and comprises up to date more than 10 billion (10x10^9) information units being gathered by an advanced meta-search engine, connecting informations being contained in thousands of scientific papers with those from the internet and with those, found in related scientific databases. Using logical rules based on findings, described in Die formatierte DNA IMPACD is able to detect coherencies and linkages not only between genes, but also between proteins and messenger-RNAs one was never before able to reveal.
The actually on this side presented informations represent the latest state of art in NCL-research: never before was given such a deep insight in the various genetic mechanisms leading to development of a specific variant of NCL.
How to read the following list:
The name of a protein followed by the number of calculated transcripts means, that a polypeptide belonging to this protein-family is supposed to be coded on this gene, but at the moment I am not able to decide, wether it is representing a precursor / polypeptidchain only or a metabolic active isoform of the respective protein. A transcript believed to represent the whole amino-acid (residue) structure of a metabolic active protein or of one of its isoforms is either followed by the number of residues and a protein ID or by two or more geneloci in brackets, designating the calculated genes, coding for the metabolic functional protein.
The following list is sorted first by chromosomes, then by geneloci in ascendent order.
CLN1/PPT1 - is coding for 1p32.2
- b-L-arabinosidase isoform (CLN1 ENST00000372775 - CLN6)
- CTSL1 isoform 3 from 8 isof. at all (CLN1-CLN5)
- CLCN7 isoform (CLN1 ENSP00000361862 -CES7)
- CLCN7 isoform (CLN1 ENSP00000361862 - DHRS7)
- palmitoyl-protein-thioesterase isoform 1 (CLN1 ENSP00000403207 - CLN3 - CES7)
- palmitoyl-protein-thioesterase isoform 5 (CLN1 ENSP00000392293 -NOMO2)
- palmitoyl-protein-thioesterase isoform 6 (CLN1 ENSP00000394863 - PPT1- 001)
- palmitoyl-protein-thioesterase isoform 12 (CLN1 ENSP00000392293 - CES7 - NOMO2)
- palmitoyl-protein-thioesterase isoform 14 (CLN1 ENSP00000392293 - DHRS7)
- palmitoyl-protein-thioesterase isoform 15 (CLN1 ENSP00000392293 - GPC6)
- protein-disulfide-isomerase isoform (CLN1 ENSP00000361862-DHRS7)
- protein-disulfide-isomerase isoform (CLN1-CES7)
- retinol-isomerase isoform 7 (CLN1 ENST00000449045- USP10 - FAM117A)
- 6 transcripts most likely not NCL related
CTSK - is coding for 1q21
- cathepsin K NP_000387
- CLCN5 - 5 transcripts
- CLCN7 - 3 transcripts
CLCN6 - is coding for 1p36
- CLCN6 8 transcripts
- CLCN2 1 transcript
- CLCN3 2 transcripts
MFSD8/CLN7 - is coding for 4q28
CLN8 is coding for (see separate table) 8p23.1
- acetoin-racemase isoform 8 (CLN8 - USP10)
- acetoin-racemase isoform 10 (CLN8 286aa - ENSP00000328182 )
- acetoin racemase isoform 11 (CLN8-CLN3
- CLCN6 isoform 9 (CLN8-DHRS7)
- CLCN7 isoform 5 (CLN8-CLN2)
- CLCN7 isoform 6 (CLN8-CES7)
- CLCN7 isoform 7 (CLN8-USP10)
- CTSL1 isoform 4 (CLN8-GPC6-CES7)
- CTSL1 isoform 5 (CLN8 - CES7)
- CTSL1 isoform 6 (CLN8-DHRS7-CES7)
- MFSD8 isoform 7 (CLN8-USP10-CLN5)
- MFSD8 isoform 8 (CLN8-DHRS7-CES7)
- MTTDC isoform 9 (CLN8-CLN3)
- PPT isoform 3 (CLN8-USP10)
- PPT isoform 7 (CLN8-CLN3)
- PPT isoform 9 (CLN8-CES7)
- PPT isoform 11 (CLN8-CLN5)
- TPP isoform 5 transcript A (CLN8-USP10)
- TPP isoform 5 transcript B (CLN8-USP10)
- TPP isoform 6 (CLN8-CES7)
I postulate CLN8P (286aa ENST00000331222) to be a precursor/polypeptidchain used to build different isoforms of above listed proteins. See separate chapter.
CTSL1 - is coding for 9q21-q22
- acetoin racemase isoform 4 (CTSL1 - CTSD)
- cathepsin L1 Var. 1 NP_001903
- cathepsin L1 Var. 2 NP_666023
- 10 other transcripts likely not NCL related
CTSL2 - is coding for 9q22.2
- cathepsin L2 NP_001324
- cathepsin L2 - 8 transcripts
- CLCN4 - 1 transcript
- FAM117A - 1 transcript
- FAM19A2 - 1 transcript
CLN2/TPP1 - is coding for 11p15.4
- CLCN6 isoform 8 (CLN2 - CES7-NOMO2)
- CLCN7 isoform 5 (CLN2 - CLN8)
- CLCN7 isoform
- CLCN7 isoform
- CTSL1 isoform
- CTSL2 isoform
- PPT isoform 16 (CLN2 ENSP00000412783 - CES7)
- protein disulfide-isomerase isoform (CLN2 - CLN5)
- tripeptidyl-peptidase isoform 7 (TPP1) ENSP00000299427
- tripeptidyl-peptidase isoform 11 ENSP00000398136
- tripeptidyl-peptidase isoform 12 (CLN2 ENSP00000395326 -CLN6)
- tripeptidyl-peptidase isoform 13 ENSP00000369169
- 10 transcripts most likely not NCL-related
CTSD/CLN10 - is coding for 11p15.5
- acetoin racemase isoform 4 (CTSD-CTSL1)
- cathepsin D isoform 5 (ENSP00000236671)
- cathepsin D isoform 7 (ENSP00000384947)
- cathepsin D isoform 3 (CTSD - DHRS7)
- cathepsin D isoform 4 (CTSD - DHRS7)
- cathepsin D isoform 9 (CTSD - CES7)
- CLCN7 isoform (CTSD - SERPINB11)
- tripeptidyl-peptidase isoform 3 (CTSD-CLN3)
- tripeptidyl-peptidase isoform 9 (CTSD-CLN3-NOMO2)
- 8 other transcripts likely not NCL related
CTSF- is coding for 11q13
- acetoin-racemase isoform 11 (CTSF - CLN3)
- cathepsin F - isoform 484aa ENSG00000174080
- cathepsin F - 5 transcripts
- cathepsin L1 - 2 transcripts
- tripeptidyl-peptidase isoform 1 (CTSF - CLN3)
- 2 other transcripts likely not NCL related
FAM19A2 - is coding for 12q14.1
CLN5 - is coding for 13q21.1-q32
- CLCN6 isoform (CLN5 - DHRS7)
- CLCN7 isoform (CLN5 - CLN6)
- CTSL1 isoform (CLN5-CLN1)
- MFSD8 isoform 7 (CLN5-CLN8-USP10)
- palmitoyl-protein-thioesterase isoform 11 (CLN5-CLN8)
- palmitoyl-protein-thioesterase isoform 13 - 407aa ENST00000377453
- protein disulfide-isomerase isoform (CLN5 - CES7)
- protein disulfide-isomerase isoform (CLN5 - CLN2)
I postulate CLN5 407aa (NP_006484) to represent isoform 13 of palmitoyl-protein thioesterase.
GPC6 - is coding for 13q32
- acetoin-racemase isoform 12 (GPC6-CLN3)
- beta-L-arabinosidase - 1 transcript
- CLCN6 isoform (GPC6 -DHRS7)
- CTSL1 isoform 4 (GPC6-CLN8-CES7)
- glypican 6 precursor 555 aa NP_005699
- MFSD8 - 1 transcript
- multivalent terpene and terpenoid decyclase isoform 8 (GPC6-CLN3-CES7)
- palmitoyl-protein-thioesterase isoform 15 (GPC6 - CLN1 ENSP00000392293)
- protein-disulfide isomerase - 1 transcript
- retinol isomerase isoform 9 (GPC6 -CLN6)
- tripeptidyl-peptidase isoform 16 (GPC6-NOMO2)
- 1 other transcript likely not NCL related
DHRS7- is coding for 14q23.1
- acetoin-racemase isoform 2 (DHRS7 - CLN3)
- cathepsin D isoform 3 (CTSD - DHRS7)
- cathepsin D isoform 4 (CTSD - DHRS7)
- CLCN6 isoform 9 (DHRS7-CLN8)
- CLCN6 isoform (DHRS7 - GPC6)
- CLCN6 isoform (DHRS7 - CLN5)
- CLCN7 isoform (DHRS7 - CLN3)
- CTSL1 isoform 6 (DHRS7-CLN8-CES7)
- dehydrogenase/reductase (SDR family) member 7 precursor 339aa
- MFSD8 isoform 8 (DHRS7-CLN8-CES7)
- multivalent terpene and terpenoid decyclase isoform 1 (DHRS7 - CLN3)
- multivalent terpene and terpenoid decyclase isoform 2 (DHRS7 - USP10 - CES7)
- multivalent terpene and terpenoid decyclase isoform 5 (DHRS7 - NOMO2)
- nodal modulator 2 isoform (DHRS7-CLN6-NOMO2)
- palmitoyl-protein-thioesterase isoform 14 (DHRS7 - CLN1)
- protein-disulfide-isomerase isoform (DHRS7 - CLN1 ENST00000449045)
- retinol-isomerase isoform 1 (DHRS7 - CLN6)
- retinol-isomerase isoform 4 (DHRS7 - CLN3 - CES7)
- retinol-isomerase isoform 5 (DHRS7 - CLN6)
- tripeptidyl-peptidase isoform 4 (DHRS7-SERPINB11)
- tripeptidyl-peptidase isoform 10 (DHRS7-CLN6-CLN3)
- tripeptidyl-peptidase isoform 15 (DHRS7-CLN6-ARSG)
CLN6 is coding for 15q23
- acetoin racemase isoform 9 (311aa ENSP00000249806)
- b-L-arabinosidase isoform (CLN1 ENST00000372775 - CLN6)
- cathepsin D isoform 2, 6, 10 (see data table)
- CLCN7 isoform 9 (CLN6 - CLN5)
- multivalent terpene and terpenoid decyclase isoforms 4 and 16 (see data table)
- nodal modulator 2 - isoform (CLN6-NOMO2-DHRS7)
- palmitoyl-protein-thioesterase isoform 2 (CLN6-NOMO2)
- retinol-isomerase isoforms 1, 5, 9, 11 (see data table)
- tripeptidyl-peptidase isoform 2 (CLN6-CLN3)
- tripeptidyl-peptidase isoform 8 (CLN6-CLN3)
- tripeptidyl-peptidase isoform 10 (CLN6-DHRS7-CLN3)
- tripeptidyl-peptidase isoform 12 (CLN6-CLN2)
- tripeptidyl-peptidase isoform 15 (CLN6-DHRS7-ARSG)
- 2 transcripts not identified, likely not NCL-realted
CLN6-protein 311aa ENSP00000249806 must be acetoin racemase isoform 9. See data table.
CLN3: see separate chapter! 16p12.1
CLCN7 - is coding for 16p13
- CLCN7 isoform a and b (NP_001107803; NP_001278.1)
- CLCN7 3 transcript
- CLCN5 3 transcripts
- CLCN4 2 transcripts
- CLCN3 2 transcripts
- CLCN2 3 transcripts
- CLCN1 5 transcripts
- CLCNKA 3 transcripts
- CLCNKB 2 transcripts
NOMO2: see separate table. 16p13.12
CES7: see separate table 16q12.2
USP10: see separate table 16q24.1
FAM18B2 is coding for 17p12
- CTSL1 - 1 transcript
- CTSL2 - 1 transcript
- FAM18B2 (family with sequence similarity 18, member B2)
- FAM18B2 - 4 transcripts
- SERPIN B11 - 3 transcripts
- and 3 other transcripts likely not NCL related
FAM117A is coding for 17q21.33
- cathepsin D isoform 8 (FAM117A-NOMO2)
- chlorid-ion channel 7 (FAM117A - USP10)
- FAM117A - NP110429.1 - 453aa
- FAM117A isoform
- GPC6 isoform (FAM117A - USP10)
- palmitoyl-protein-thioesterase isoform 10 (FAM117A - USP10)
- retinol-isomerase isoforms 3, 7 and 10 (see data table)
ARSG is coding for 17q24.2
- arylsulfatase G precursor NP_055775.2
- CLCN7 isoform (ARSG - MFSD8)
- PPT isoform 8 (ARSG - USP10 - CLN3)
- tripeptidyl-peptidase isoform 15 (ARSG-CLN6-DHRS7)
- and 8 other transcripts likely not NCL related
SGSH is coding for 17q25.3
- CC-preferring endodeoxyribonuclease EC 3.1.21.6 - 2 transcripts
- chlorid-ion channel 7- 1 transcript
- N-sulfoglucosamine sulfohydrolase NP_000190
- N-sulfoglucosamine sulfohydrolase isoforms - 4 transcripts
- protein of glypican family - 2 transcripts
- protein of family with sequence similarity 18, member B2 - 1 transcript
- protein of family with sequence similarity 19 member A2 - 1 transcript
- and 5 other transcripts likely not NCL related
SERPINB11 is coding for 18q21
- acetoin racemase isoform 10 (SERPIN B11 - USP10)
- cathepsin D isoform 1 (MFSD8 - SERPINB11)
- CLCN7 isoform (SERPINB11 - CTSD)
- multivalent terpene and terpenoid decyclase isoform 14 (SERPINB11 - USP10 )
- serpin peptidase inhibitor, clade B, member 11 (ENSP00000421854) - 5 transcripts
- tripeptidyl-peptidase isoform 4 (SERPINB11-DHRS7)
- 6 other transcripts likely not NCL-related
Discussing geneloci CLN1, CLN3, CLN4, CLN5, CLN6, CLN7, CLN8, CLN9 and CLN10.
Preliminary remarks:
The following chapters discussing particular variants of NCL are from case to case equipped with tables listing expression data for single proteins. These data should be interpreted as timeframes showing the overall expression period for a distinct isoform of a protein. That is to say, that expression time for that protein in a specific cell may be different within the borders set by the documented timeframe, because a protein consisting of a distinct polypeptidchain may have been generated from two or more different transcripts, them only differing in the length of 5 and/or 3 noncoding areas. Vice versa this further means, that the number of transcripts generated from this gene must not be identic with the number of different proteins, postulated to be encoded by the specific gene.
Usage of terminus polypeptide in scientific literature and in www is not consistent. In this work polypeptide or polypeptidechain will characterise a distinct number of linked aminoacids, which are used in connection with a polypeptidechain (or chains) generated from other genes to build isoforms of proteins involved in metabolism of the cell.
NCL-diseases CLN1 through CLN10 will now - with exception of CLN2 - be discussed in detail:
CLN1, usually defined as infantile NCL, is a multifactorial and in 40% of cases a multigenetic disease too.
CLN1 or Santavuori-Haltia type or infantile NCL is believed to be caused by a missing or defective isoform of palmitoyl-protein-thioesterase 1 (PPT1) - a small glycoprotein. PPT1 is thought to be involved in catabolisme of lipid-modified proteins (Camp et al., 1994) by removing palmitate groups from cysteine residues from those proteins.
Human PPT1 was first characterisised by Schriner et al. (1996) - them predicting a 306-amino acid polypeptide that contains a 25-amino acid signal peptide, 3 N-linked glycosylation sites, and consensus motifs distinctive of thioesterases. Northern blot analysis revealed ubiquitous expression of a single 2.5-kb mRNA, with highest expression in lung, brain and heart.
From OMIM: Heinonen et al. (2000) analyzed the intracellular processing and localization of adenovirus-mediated Ppt in mouse primary neurons and in nerve growth factor (see 162030)-induced PC12 cells. The neuronal processing of Ppt was found to be similar to that observed in peripheral cells, and a significant amount of the PPT enzyme was secreted in the primary neurons. Immunofluorescence analysis of the neuronal cells infected with wildtype Ppt showed a granular staining pattern in the cell soma and neuronal shafts. Interestingly, Ppt was also found in the synaptic ends of the neuronal cells, and the staining pattern of the enzyme colocalized to a significant extent with the synaptic markers SV2 (185860) and synaptophysin (313475). Heinonen et al. (2000) found that their in vitro data corresponded with the distribution of endogenous Ppt in mouse brain and suggested that Ppt may not solely be a lysosomal hydrolase. Heinonen et al. (2000) suggested that the specific targeting of Ppt into the neuritic shafts and nerve terminals indicates that Ppt may be associated with the maintenance of synaptic function, and speculated that the enzyme could have an extracellular substrate as well. (end of cit.)
To the same conclusion came Lehtovirta et al. (2001), which determined the neuronal localization of PPT1 by confocal microscopy, cryoimmunoelectron microscopy and cell fractionation. In mouse primary neurons and brain tissue, they detected PPT1 in synaptosomes and synaptic vesicles but not in lysosomes. In polarized epithelial Caco-2 cells PPT1 was localized exclusively in the basolateral site, in contrast to the classic lysosomal enzyme aspartylglucosaminidase (AGA; 613228), which is localized in the apical site. Lehtovirta et al. therefore hypothesized that PPT1 also might play a role outside of lysosomes in the brain and may be associated with synaptic functioning.
According to my computational calculations based on IMPACD® gene CLN1/PPT1 is coding 19 polypeptids, from which 6 are based on my data likely not NCL-related. The remaining 13 polypeptides are used to generate isoforms for 6 different protein-families as listed below:
As I have mentioned several times before and in Die formatierte DNA a given protein may be encoded by several different transcripts. This is according to my data also the case with common CLN1/PPT1 protein: the manually curated ENSEMBLE-transcript ENST00000433473 consisting of 2742 basepairs coding the 306 aa protein PPT1 will be expressed based on my data solely during puberty. A second transcript encoding an identic 306 aa protein which I have calculated with 1551 basepairs will be generated from birth until reaching adult age, while a third transcript with 306 aa documented in my data will be likely synthesised from birth until around age 35 and counts 1950 baisepairs.
These three different transcripts encoding the same 306 aa protein, actually known as PPT1 will be doubtlessly expressed - likely with different modifications - in different celltypes - in which I will verify possibly later - and it should be obvious, that the numerous mutations in CLN1 must have different effects on expression of these 3 transcripts and naturally on various other transcripts, generated from CLN1 during ontogenesis.
Supposed this being correct, PPT1 solely for that reason can not be the main or the single cause of CLN1, especially in regard to the different phenotypes of CLN1, which show not only different onsets, but also very different courses of disease. Any other deduction would be illogical and inconsequent.
Nine different proteins most likely will be involved in CLN1 disease.
Computational expression analysis for gene CLN1 shows 6 different polypeptides used to build isoforms of PPT, cells thereby coupling polypeptides coded on gene CLN1 with those coded on genes like DHRS7, CES7, GPC6, CLN3 and NOMO2 to generate functional proteins. Four of these PPT-isoforms (5,12,14,15) use polypeptide ENSP00000392293 with 203 residues in combination with peptides coded on genes CES7, DHRS7, GPC6 and NOMO2 to build functional proteins.
Attention we should direct also on those two isoforms of chlorid-ion-channel-7 protein, which will be generated according to my data by linkage of the 232-residues-polypeptid ENSP00000361862 with a polypeptid coded on gene CES7 respective DHRS7 and on isoform 7 of retinol isomerase, encoded by polypeptides generated from genes CLN1, USP10 and FAM117A.
A compilation of expression data (sorted by begin of expression) for those transcripts/proteins/isoforms I assume to cause CLN1 shows the following expression periods.
|
PPT-1
(CLN1ENSP00000403207- CLN3 -CES7)
|
from day 110 till birth
|
|
PPT-5
(CLN1ENSP00000392293 - NOMO2)
|
from birth till end of year 5
|
|
PPT- 6
(PPT1 ENSP00000394863)
|
from birth till around age 35 (3 diff. transcr.)
|
|
retinol-isomerase 7
(CLN1 - USP10 - FAM117A)
|
from end of y5 till begin of puberty
|
|
CLCN7 isoform
(CLN1ENSP00000361862- CES7)*
|
from begin of puberty till begin of adultness
|
|
CLCN7 isoform
(CLN1ENSP00000361862- DHRS7)*
|
from begin of puberty till begin of adultness
|
|
PPT-12
(CLN1ENSP00000392293 - CES7 - NOMO2)
|
from begin of puberty till begin of adultness
|
|
PPT-14
(CLN1ENSP00000392293 - DHRS7)
|
from begin of puberty till ~year 35
|
|
PPT-15
(CLN1ENSP00000392293 - GPC6)
|
from begin of puberty till end of life
|
|
* The two isoforms of CLCN7 synthesised within identic timeframes
will be expressed in different cells.
|
|
More than 48 different mutations on CLN1/PPT1 spread over the whole gene will affect without any doubt not only PPT1.
CLN1 disease becomes apparent in at least 4 different phenotypes. If one would be able to verify the impact of a specific mutation on the expression of one or more of the above listed proteins, we would count as a matter of fact most likely more than 10 variants only for CLN1.
And in most - if not all - of these variants there would be more than one protein affected by a distinct mutation in CLN1. Therefore CLN1 should be designated as a multifactorial disease, as the most of NCLs according to the results of this most comprehensive study of NCL-causing genes ever should be designated too.
Roughly 40% of CLN1-diseases must be associated with another NCL-related mutation wether on gene DHRS7 or on gene CES7.
Analysing the phylogenetic inheritance of genes belonging to the NCL-genes-and-protein network it has very high probability, that around 20% of patients suffering on a CLN1 mutation are also affected by a NCL-related mutation on DHRS7 and another approx. 20% by such mutation on CES7.
A summary of proteins coded on DHRS7 may be seen here, and this is where I give an overview on CES7.
This coupling of mutations on different genes will surely further contribute to the inhomogenous clinical presentation of this type of NCL and will be found by other variants of this disease too.
Cellular localisation of PPT1 will be defined by posttranslational modifcations.
From PPT1 306aa (in list above designated as isoform 6 according to my system of numeration - see table) I postulate to exist 7 different trafficking or transportation variants, which will be generated by postranslational modifications at specific sites of the protein. In this regard PPT1 is much resembling CLN3P/MTTDC, the cellular trafficking of which is depending from posttranslational modifications too. This will be discussed further down on this side.
PPT1 is known to be ubiquitiously expressed within different cells (likely in around 30 different cell populations), and in these cells it has been found in different compartments respective different organells. PPT1 being well known to be found in lysosomes of various cells for a considerable time, Heinonen located Ppt1 in the synaptic ends of neuronal cells, while Lehtovirta found PPT in synaptosomes and synaptic vesicles in mouse primary neurons and brain tissue, but not in lysosomes of those cells.
I postulate PPT1 (PPT-6) to be trafficked depending from specific modification(s) to 7 different targets in also different celltypes:
endosomes ->lysosomes
membranes of synaptic vesicles
synaptic bodies (synaptic ribbons)
nuclear membranes
membranes of mitochondriae
membranes of ER
membranes of Golgi-apparatus
With exception of synaptic bodies/synaptic ribbons all above listed organells are well known, their function is well-investigated too. In contrast to that, function of synaptic bodies/synaptic ribbons is widely unknown or in discussion.
Cited from electron microscopic atlas edited by H. Jastrow:
Synaptic bodies (SB; Terminologia histologica: Corpuscula synaptica) consist of an electron-dense proteinaceous structure that is surrounded by synaptic vesicles. In most cases SBs appear as synaptic ribbons (Terminologia histologica: Fasciolus synapticus). They are located close to the cell membrane or attached to the latter close to the active zone where neurotransmitter is released, i.e. they are presynaptic.
SB are encountered in synapses with a high vesicle turnover: photoreceptor cells (rods and cones) and bipolar cells of the retina, cochlear receptor cells of the inner ear (inner and outer hair cells; here often spheres or irregular lump-like structures), receptor cells of the vestibular organs (Macula sacculi and utriculi as well as the Crista ampullaris: bottle-like hair cells [type 1 cells] and cylindrical hair cells [type 2 cells]), celles of the paratympanic organ and pinealocytes, the parenchymal cells of the pineal gland.
SB bind glutamate-containing neurotransmitter vesicles* via fine proteinaceous "arms". They change in morphology and size depending on light conditions and stimuli and constitute a reservoir of synaptic vesicles. Some autors assume that they transport synaptic vesicles to the cell membrane. (end of cit. - *emphasis added)
Besides PPT also MTTDC, AR, TPP, CTSL1 and CLCN7 should be found in synaptic bodies and in membranes of synaptic vesicles.
My own calculations and evaluations based on analysis of concerted expression of distinct gene groups let me postulate, that besides PPT also MTTDC, AR, TPP, CTSL1 and CLCN7 have to be found in synaptic bodies as well as in membranes of synaptic vesicles - synaptic bodies in my opinion representing a kind of support base for the latter. Additionally to these 6 proteins postulated to be found in synaptic vesicles and -bodies most likely there will be detected CTSL2 and CTSF in synaptic bodies too.
Also endosomes/lysosomes in all stages of development I postulate to harbour the previously mentioned eight proteins and besides those most likely CTSD, CTSK, CLCN6, PDI and beta-L-arabinosidase - them all belonging to the NCL-Protein-Network. This may be one of the reasons, why certain variants of NCL linked with a malfunction of endosomes/lysosomes present themselves of a similar phenotype, despite the genetic roots being quite different.
CLN2 analysis is in progress........
CLN2-disease will depending from resources possibly be analysed and discussed at a later time.
CLN3 or classical juvenile and adult NCLs.
Mutations on gene CLN3 being the most abundant cause for getting NCL this type of disease will be discussed in detail at the end of this side.
CLN4: adult NCLs and Kufs disease.
Main cause are mutations on genes GPC6, DHRS7 or CES7.
Actually adult NCL or Kufs disease is brought in connection with gene CLN1, more precisely with mutation cln1.044 and cln1.050 in that gene.
Using an statistical approach and looking at the above shown genetic pedigree, the overview of NCL-related genes and the data tables for MTTDC, PPT, TPP, AR, RI and CTSD we will soon realise that it may not be possible to causally associate the adult variants of NCL - also called Kufs disease - with a single gene or a single gene-product.
Not only for that reason it seems very unlikely gene CLN1 being the single or solely cause for CLN4: according to my data most likely six different genes are crucially involved in aetiology of different phenotypes of adult NCL, if we define adult NCL by an onset after the twentieth year of life35. These six genes are (in alphabetical order): CES7, CLN1, DHRS7, GPC6, NOMO2 and USP10. This statement refers to CLN4A (the recessive inherited form) as well as to CLN4B (the dominant form).
From these before mentioned genes predominantly GPC6, DHRS7 and CES7 are causally involved in adult NCL-variants: trying to get statistical values for a correlation between one of these genes and CLN4 - based on phylogenetic inheritance - we should differ those cases caused by a single mutation on one of these genes from those with linked mutations on two different genes, the latter variant most likely being very often the case with CLN4-disease.
A single gene defect on GPC6 by example should be present in 40% of all CLN4-cases, including a combination of a GPC6-mutation with such on CES7 will increase this value to around 55%, including also combinations of defects on GPC6 and DHRS7 will further enlarge this figure to 60%.
Cause of different variants of CLN4 must be mutations on previously mentioned genes, involvement of a distinct gene may be statistically calculated as listed below (appr. values, sum > 100 because of combined mutations):
|
GPC6
|
40%
|
|
DHRS7
|
40%
|
|
CES7
|
20%
|
|
CLN1
|
5%
|
|
USP10
|
5%
|
|
NOMO2
|
5%
|
Linked mutations on two different genes will appear between all of the above mentioned genes including CLN1: approximately 25% of CLN4-patients with a mutation on GPC6 should have an defect on CES7 too - more 20% a genetic defect on GPC6 and DHRS7.
Other combinations of genetic defects should occurr between genes NOMO2 and USP10, but those will surely play a minor role, if we relate minor to the percentage of cases and not to the severity of the individual disease.
The most (~60 %) of the adult variants of NCL therefore must be caused by different mutations on gene GPC6, case-by-case linked with a mutation on a second gene of the above defined gene-array.
Looking at the analysis of genes GPC6, DHRS7 and CES7 we will note some well known proteins being encoded on these loci and some unknown to be related to NCL postulated by the author. Depending from distinct mutation(s) on one or more of the above defined genes, a specific variant of adult NCL may therefore not only be connected with distinct defective or missing isoforms of palmitoyl-protein thioesterase, but also induced by defective or missing isoforms of acetoin-racemase, multivalent terpene and terpenoid decyclase, tripeptidyl-peptidase, protein-disulfide isomerase, MFSD8, retinol isomerase, chlorid-ion channel 6 protein or any other protein being encoded on those genes, depending from connection with an individual mutation.
Isoforms of the previously mentioned protein-families all being emerged from a common progenitor as shown in genetic pedigree and mostly linked on the molecular level as shown in data tables and in overview of NCL-related genes I hereby postulate to underly the various phenotypes of adult NCLs.
My data show clearly without ambiguity, that the two by Berkovic, Carpenter et al. former defined phenotypes of Kufs disease8 and all other uptodate documented cases of NCLs with an onset in adult age cannot be caused by a single defective protein, but must be induced by two, three or even more defective isoforms of a protein encoded on one or more of the above defined genes. Therefore a clear and distinct differentiation between type A and type B of Kufs disease is according to my findings impossible. This also is the final conclusion of Vadlamudi et al. after reviewing 5 patients with biopsy-proven Kufs disease and 14 case reports of Kufs disease taken from literature. They write:
[..]Conclusions: The inheritance, mechanism, and manifestations of Kufs disease are not well understood. EEG findings may guide clinicians toward a confirmatory pathological diagnosis and distinguish various phenotypes of this disorder.9(end of cit.)
Looking at my data tables and realising the expression pattern for the CLN4-causing genes it should be apparent, that some adult forms of NCL could have an undetected or perhaps unremarkable (or unremarked) onset at earlier stages before adulthood. This statement in a transferred sense may be valid for the most of the various forms of NCL too.
|
CLN5 - the so called finnish variant of late infantile NCL has most likely a multifactorial as well as a multigenetic basis.
From OMIM: CLN5 was classically described in Finnish patients with onset between 4 and 7 years of age and is often referred to as the 'Finnish variant of late-infantile NCL' (Finnish vLINCL). With the identification of molecular defects, however, the CLNs are now classified numerically according to the underlying gene defect. CLN5 refers to CLN caused by mutation in the CLN5 gene, regardless of the age at onset.
Symptoms of CLN5 are reported by C.H. Chang as follows:
Onset at age 4.5-7 years
Motor clumsiness
Concentration problems
Similar to CLN2 but slower course
Death in second or third decade
Gene CLN5 is according to my calculations coding 9 polypeptides, used to synthesise isoforms of proteins belonging to 7 different families. CLN5P - a protein of 407 residues believed to be main cause of this variant of NCL - must according to my evaluations be an isoform of PPT.
The gene is coding in detail for:
acetoin racemase isoform 4 (CLN5 - GPC6)
CLCN6 isoform (CLN5 - DHRS7)
CLCN7 isoform (CLN5 - CLN6)
CTSL1 isoform (CLN5-CLN1)
MFSD8 isoform 7 (CLN5-CLN8-USP10)
palmitoyl-protein-thioesterase isoform 11 (CLN5-CLN8)
palmitoyl-protein-thioesterase isoform 13 - 407aa ENSP00000366673
protein disulfide-isomerase isoform 6 (CLN5 - CES7)
protein disulfide-isomerase isoform 7 (CLN5 - CLN2)
Regarding expression data for this gene the circumstances therefore are similar to those of the whole group of NCL-causing genes, none of these genes only coding for a single transcript or protein, but for a group of NCL related- or NCL-causing proteins.
To fix CLN5-disease to a certain protein or to certain transcripts or proteins will be as long impossible, as this fixing will not be linked with a distinct mutation. But in general it is highly unlikely, that any mutation on CLN5 (showing only 4 exons) will be restricted to a single transcript or polypeptide.
Up till now 27 mutations have been found in CLN5 - spread all over the whole gene. One of the most abundant in the finnish population - mutation cln5.0001 (OMIM) - cln5.001 in NCL resource database, a two basepairs deletion in exon 4 - is generating a predicted truncated protein of 391 residues, which I postulate to have no residual function and therefore should be eliminated by internal cellular control processes.
According to my computational analysis mutation cln5.0001 affects most likely the following 5 proteins:
palmitoyl-protein-thioesterase isoform 11 (CLN5 - CLN8)
protein disulfide-isomerase isoform 6 (CLN5 - CES7)
protein disulfide-isomerase isoform 7 CLN5 - CLN2)
CLCN6 isoform (CLN5 - DHRS7)
CTSL1 isoform (CLN5 - CLN1)
But these proteins partially coded on CLN5 may not be the main or solely cause for the different CLN5-variants.
Any NCL-related mutation on gene CLN5 must be coupled with a mutation wether on gene USP10 or on CLN6.
Gene CLN5 being derived from USP10, this gene being arised from a duplication of CLN6, I postulate the whole group of any CLN5-mutation bearing patients to suffer wether on a second mutation on USP10 (~60%) or on such defect on CLN6 (~40%).
The specific phenotype of CLN5-disease therefore is depending from the specific combination of mutations on gene CLN5 and gene CLN6, resp. on gene CLN5 and gene USP10. USP10 as well as CLN6 themselves encoding various members of the NCL-protein-family, the distinct phenotype of any CLN5-related NCL-variant therefore is depending from the actual mutation(s) and their impact on the expression of proteins coded on these genes.
This being verified, CLN5 should be renamed to CLN5/CLN6 respective CLN5/USP10.
The up to date undefined CLN6-protein of 311 residues must be an isoform of acetoin racemase.
Very little is known about CLN6-disease, which was classically described as a late infantile or juvenile onset form, with onset between 2 and 6 years of age, occurring outside of the Finnish population. CLN6 was often referred to as a variant of late-infantile NCL (vLINCL). Celia H. Chang characterises the disease as following:
CLN6 or variant late infantile/early juvenile NCL (Lake Cavanagh disease)
Onset between age 18 months to 8 years
Visual loss
Seizures
Resembles CLN2
Loss of motor skills between age 4-10 years
Death in the second or third decade
Sara Mole (NCL resource) in contrast to this writes: CLN6 - Mutations generally cause NCL with onset in late infancy.
Till today (january 2011) 46 different mutations have been identified resulting in missense codons, aberrant splicing, frameshifts, deletions, extra amino acids and other irregular expression patterns. Mutations are spread over all exons and will be found also in some introns causing by example aberrant splicing.
According to my calculations and evaluation of my data from gene CLN6 during ontogenesis will be generated 25 different transcripts, which are coding 14 different polypeptides, them being combinated with peptides encoded by other genes to form 19 distinct isoforms of previously defined NCL-related proteins as shown below:
acetoin racemase isoform 9 (CLN6-001 ENSP00000249806 311aa)
b-L-arabinosidase isoform (CLN6 - CLN1/PPT1-004 ENST00000372775)
cathepsin D isoform 2 (CLN6 - NOMO2)
cathepsin D isoform 6 (CLN6 - CLN3)
cathepsin D isoform 10 (CLN6 - CLN3)
CLCN7 isoform 9 (CLN6 - CLN5)
multivalent terpene and terpenoid decyclase isoform 4 (CLN6 - CLN3 see data table)
multivalent terpene and terpenoid decyclase isoform 16 (CLN6 - CLN3)
nodal modulator 2 - isoform (CLN6-NOMO2-DHRS7)
palmitoyl-protein-thioesterase isoform 2 (CLN6-NOMO2)
retinol-isomerase isoform 1 (CLN6 - DHRS7 see data table)
retinol-isomerase isoform 5 (CLN6 - DHRS7)
retinol-isomerase isoform 9 (CLN6 - GPC6)
retinol-isomerase isoform 11 (CLN6 - NOMO2)
tripeptidyl-peptidase isoform 2 (CLN6-CLN3)
tripeptidyl-peptidase isoform 8 (CLN6-CLN3)
tripeptidyl-peptidase isoform 10 (CLN6-DHRS7-CLN3)
tripeptidyl-peptidase isoform 12 (CLN6-CLN2)
tripeptidyl-peptidase isoform 15 (CLN6-DHRS7-ARSG)
My data show clear and without ambiguity, that CLN6-protein 311aa ENSP00000249806 - besides representing an metabolic functional isoform of acetoin racemase - is acting also as a precursor / polypeptidechain for building eleven more different proteins (in list above marked red) and in this way showing once more that there is in fact an integrated network of genes and proteins underlying the various forms of NCL.
It should be obvious that the different mutations mentioned before in most cases will affect also different proteins - and very often a given mutation will moreover modify or disable expression or synthesis of more than one protein, so giving cause for the reported inhomogenous onset and variant progression of this type of NCL.
Looking at the data tables and the expression data documented therein we should remember, that listed expression times for isoforms of proteins have to be interpreted as timeframes setting the borders for expression of different transcripts with also different expression times, which encode the same isoform.
CLN6-protein with 311 residues I have identified as isoform 9 of acetoin racemase, one of the key enzymes of butanoate metabolisme, which itself is linked with
Glycolysis / Gluconeogenesis
Citrate cycle (TCA cycle, Krebs cycle)
Synthesis and degradation of ketone bodies
Alanine, aspartate and glutamate metabolism
Pyruvate metabolism
Vitamin B6 metabolism
Biosynthesis of type II polyketide backbone
Expression data retained by computational analysis via IMPACD® show CLN6P/acetoin racemase 9 will be encoded during ontogenesis by 5 different transcripts which will be generated within 5 different periods, in detail:
from birth until end of year 5
from end of year 5 until begin of puberty
from begin of puberty until reaching adultness
during puberty
from begin of puberty until end of life
CLN6P/acetoin racemase 9 is according to my calculations predominantly expressed in neurons, in cells of oligodendroglia (please read this lines), muscle cells (smooth and striated), lymphocytes, specialised cells of the retina (amacrine-cells and Müller-cells) and in many celltypes of liver and kidneys.
Discussing CLN1 I have postulated acetoin racemase to be part of synaptic bodies and to be resident in membranes of synaptic vesicles. Within endosomes and all stages of lysosomes I assumed acetoin racemase together with proteins beta-L-arabinosidase, CLCN6, CLCN7, CTSD, CTSL1, CTSK, MTTDC, PDI, PPT and TPP to be found too.
But in spite of that CLN6P/acetoin racemase 9 can not be the solely cause for CLN6 - most of proteins above listed, if not all - will be linked to a more or less large number of mutations listed in databases, so contributing to inhomogenous manifestation of this disease.
Taking all these statements together and regarding the expression data for acetoin racemase 9 we may find convincing explanations for onset and progression of CLN6 disease, which - as other NCLs discussed before - should also be apostrophised as a multifactorial and multigenetic disease, due to a coupling of mutations on CLN6 with those on DHRS7 (~30% of CLN6- cases) and NOMO2 (~10% of cases).
Gene CLN7 is coding proteins containing transporter-domains for proteins belonging to NCL-gene-and-protein network.
Gene CLN7/MFSD8 among others is coding a protein called major facilitator superfamily domain containing 8 (MFSD8), which is thought to belong to a group of transporter proteins. Mutations in this gene are brought in close relationship with late infantile NCL. It is commonly suggested, that function of MFSD8 as transporter protein is causative for late infantile NCL. But until today one was not able to identify any protein, which could serve as substrat for the postulated function of MFSD8.
As already shown above I actually have calculated seven polypeptides to be generated from this locus:
I postulate MFSD8 isoform 473aa (ENSP00000425000) to contain four transporter-domains for specific isoforms of retinol isomerase, PPT, TTP and acetoin racemase - protein 518aa (ENSP00000296468) to contain 7 transporter domain(s) - from those six for specific isoforms of multivalent terpene and terpenoid decyclase and one for a specific isoform of TPP. Other transporter proteins coded on this locus most likely contain such domain(s) for other isoforms of same proteins or may contain such domains for isoforms of other NCL-related proteins as by example acetoin-racemase, PPT and others.
These findings show once more, that all genes and proteins resumed in this work are building a wide-spanning, but distinct network as revealed on this side.
MFSD-proteins in this regard can be characterised as unspecific members of the NCL-familiy, them not being part of an enzymatic pathway and also not representing an enzyme responsible for degradation or elimination of cell waste, but being proteins, whose task it is to transport members of the NCL-family with those before mentioned assignments to their related workplaces. MFSD8 protein being closely linked to other NCL-related proteins it may not be unexpected that the coding gene is a member of the phylogenetic tree of NCL-related genes too.
Besides those mentioned transporter proteins most likely a precursor for a cathepsin D isoform with relevance to late juvenile NCL, whose completing second polypeptidchain is encoded on gene SERPINB11, will be generated from gene MFSD8. This isoform will be active from around day 160 until birth and may be found in nerve-cells, striated and smooth muscle cells, lymphocytes and cells of oligodendroglia, microglia and others.
|
CLN8-disease, Northern epilepsy and the so-called
turkish variant of late infantile NCL.
Cited from NCBI: This gene encodes a transmembrane protein belonging to a family of proteins containing TLC domains, which are postulated to function in lipid synthesis, transport, or sensing. The protein localizes to the endoplasmic reticulum (ER), and may recycle between the ER and ER-Golgi intermediate compartment. Mutations in this gene are associated with progressive epilepsy with mental retardation (EMPR), which is a subtype of neuronal ceroid lipofuscinoses (NCL). Patients with mutations in this gene have altered levels of sphingolipid and phospholipids in the brain (end of cit.).
CLN8 gene, which is positioned on chromosome 8p23.1, is according to my calculations coding 25 transcripts, representing 14 different polypeptides used to build 19 different proteins, which I suppose to be expressed in different cells and within different times during ontogenesis as documented in documents below. All transcripts except that coding acetoin racemase 10 (CLN8P 286aa) will be combinated with transcripts/polypeptidechains from other genes to generate isoforms of proteins belonging to the NCL-protein-network as shown in table and diagram.
According to my computational analysis CLN8P will be used to construct distinct isoforms of CLCN6-protein, MTTDC, PPT and TPP (in table marked with d).
|
|
|
|
Oligodendrocytes mentioned on this website refers to a group of specialised cells, which resemble - even they are according to my evaluations descended from a common stem cell - not very much in their form and appearance and still less in relation to their function. One of these cells is the oligodendrocyte, which is building the myelin sheath of the neuronal axon in the central nervous system (brain and spinal cord) of higher vertebrates - a function, which is performed by Schwann cells in the peripheral nervous system.
The other one is not very well evaluated and its function is in large parts unclear. The so called satellite oligodendrocytes are always found in the neighbourhood of neurons, to which they have lot of contacts via their cellular branches. I suppose satellite oligodendrocytes to have a function similar to nurse cells - they help to regulate the content of Natrium and Calcium ions, supply the neurons with glykogen in case of high consumption and may also deliver neurotransmitters like glutamate or acetylcholin if needed. They in this way most likely form a kind of emergency troop, helping neurons to keep up their function and to survive even when there is a serious lack of nourishment, by example under stress.
|
|
CLN8 gene is suspected to cause at least two different phenotypes of NCL: the Turkish variant of late infantile NCL and the so called Northern Epilepsy, the latter being predominantly present in Finland. The follwing five paragraphs are excerpts based on informations taken from OMIM by NCBI:
Hirvasniemi et al. (1994)38 documented genealogic and phenotypic features of a recessively inherited form of childhood epilepsy occurring in the population of northern Finland, referred to as 'Northern epilepsy.' With one exception, both parents of all eleven sibships with affected individuals descended from one or two founding couples. The patients were normal at birth and developed normally until school age. Age at onset ranged from five to ten years (mean, 6.7 years) with generalized tonic-clonic seizures. The seizures increased in frequency reaching a maximum of approximately one or two seizures per week by puberty. After puberty, the frequency of seizures began to decline unrelated to changes in medication.
In early adulthood frequency of seizures was between six and twentyfive attacks per year, and after thirtyfive years of age many patients were virtually seizure-free. EEG showed focal and nonfocal paroxysmal seizures. Mental development, which was originally normal, began to deteriorate two to five years after the onset of epilepsy, and the deterioration continued during adulthood in spite of good epilepsy control, leading to mental retardation by middle age.
Haltia et al. (1999) and Herva et al. (2000) recognized Northern epilepsy as a subtype of neuronal ceroid lipofuscinosis. Herva et al. reported neuropathologic findings of 3 patients with Northern epilepsy. There was intraneuronal accumulation of cytoplasmic autofluorescent granules that were immunoreactive to subunit C of mitochondrial ATP synthase. Membrane-bound storage cytosomes showed a curvilinear ultrastructure with admixture of some granular components. The findings confirmed Northern epilepsy as a form of CLN with an exceptionally protracted course.
Ranta et al. (1999)44 found that 22 Finnish patients with the Northern epilepsy variant of CLN8 were homozygous for an arg24-to-gly mutation in the CLN8 gene. In the NCL resource database, this mutation got the number cln8.001. As mentioned before, the findings indicated a founder effect based on one or two founding couples.
Ranta et al. (2004)39 noted that although Northern epilepsy is allelic to CLN8, the clinical phenotype is distinct. Northern epilepsy presents between 5 and 10 years of age with frequent tonic-clonic seizures followed by progressive mental retardation. Visual loss is not a prominent feature of Northern epilepsy, there is no myoclonus, and the clinical progression is slower.
To make the confusion more complete, the so called turkish variant of late juvenile NCL (vLINCL)- in the meantime renamed from CLN7 to CLN8 - whose phenotype had been considered a distinct clinical and genetic entity among the NCLs - was detected not only in Turkey, but also in Italy, Pakistan and Israel and even in Germany.
Ranta et al. reported homozygosity over the Northern epilepsy CLN8 gene region on 8p23 in four out of five Turkish vLINCL families studied. However, no common mutation in CLN8 was found in these families. Later they extended the Turkish vLINCL family panel to 18 families, of which only one was nonconsanguineous. Nine families were excluded from CLN8 by lack of homozygosity. In the remaining families, four CLN8 gene mutations were identified indicating that in a subset of patients with Turkish vLINCL the disorder is allelic to Northern epilepsy. But there was no apparent genotype-phenotype correlation among the Turkish patients with CLN8 mutations, although their phenotype was distinct from that of Finnish Northern epilepsy patients.
Ranta et al. close their study with the commentary, that the molecular genetic background of the Turkish vLINCL families not linked to CLN8 remains to be clarified.
I will make an effort to shed light upon the CLN8-problem.
The real cause of northern epilepsy .
Up to date (january 2011) the NCL mutation database shows 16 disease causing mutations for gene CLN8. From these 16 mutations cln8.001 is believed to underly northern epilepsy as described by Hirvasniemi et al. (1994) based on 10 affected families in Finland. Analysing and studying (using IMPACD®) the consequences of cln8.001 mutation I have come to the strict conclusion, that gene CLN8 can not be the main or solely reason for northern epilepsy, if linked with mutation cln8.001. Why not?
In (compressed) details: gene CLN8 likely contains 3 exons - the subdomain containing this gene shows 3 promotors and 4 terminators (poly-A-signals) for this gene. Mutation cln8.001 is resident in exon 2 of transcript ENST00000331222. Calculating the 3 hnRNAs and the generated/spliced mRNAs from promotor 2 the potential negative effect of mutation CLN8.001 in exon 2 must be restricted to 8 transcripts at all, representing the following 7 distinct proteins:
- AR isoform 10 identic with CLN8P 286aa
- AR isoform 11 coded on genes CLN8 - CLN3
- CLCN6 isoform 9 coded on genes CLN8 - DHRS7
- CLCN7 isoform 6 coded on genes CLN8 - CES7
- MTTDC isoform 9 coded on genes CLN8 - CLN3
- PPT isoform 7 coded on genes CLN8 - CLN3
- TPP isoform 5 coded on genes CLN8 - USP10 (two diff. transcripts)
Others of above listed polypeptides encoded by gene CLN8 according to my data will never use this (part of) exon 2.
Looking at the expression scheme (diagram) for mutation-cln8.001 affected proteins we see, that those proteins, which I postulate to be defective, will be generated between birth and begin of adultness within various periods.
Taking the observations of Hirvasniemi et al. for granted it is obvious, that the expression scheme of above listed 7 proteins (eight transcripts) will even then merely be compatible with pathogenesis of northern epilepsy as described by Hirvasniemi et al., if we presume an retarded or protracted onset of the disease based on defective proteins with reduced functionality or activity. Since under this condition the further development of northern epilepsy - mainly after reaching adultness - will remain unexplicable, if we postulate northern epilepsy solely to be caused by mutation cln8.001.
To detect the reason for this contradiction to my findings I have used a prediction model implemented in IMPACD® to prognosticate the impairment of functionality resp. enzymatic activity of above listed 7 proteins due to mutation cln8.001.
These were the results:
|
Protein affected by mutation cln8.001
|
Remaining functionality or activity of the distinct protein (appr.)
|
|
AR isoform 10 (CLN8P 286aa)
|
50%
|
|
AR isoform 11
|
50%
|
|
CLCN6 isoform 9
|
50%
|
|
CLCN7 isoform 6
|
50%
|
|
MTTDC isoform 9
|
50%
|
|
PPT isoform 7
|
50%
|
|
TPP isoform 5
|
50%
|
|
Though these data were obtained using a prediction model it may be much likely them being correct, if we regard the character of mutation cln8.001 (missense mutation c.70C>G), which is leading to a change from arginine to glycine. Or to say it in other words: if the reduction of activity or functionality of proteins AR, TPP, PPT and MTTDC - them all being activated just at birth - would be greater than above calculated, onset of disease ought to be at birth or shortly after birth and not at age 3 to 7.5 years (turkish variant) respective after reaching school age (northern epilepsy) or even later. We remember:
The patients were normal at birth and developed normally until school age. Age at onset ranged from five to ten years (mean, 6.7 years) with generalized tonic-clonic seizures.
Mutation cln8.001 can neither be the main nor the solely cause for Northern Epilepsy nor for turkish or other variants of LINCL.
Though I am wether a paediatrician nor a clinician, I however dare to postulate, that mutation cln8.001, identified as the disease-causing mutation by Ranta et al. (1999), can not be the main or solely cause for northern epilepsy as described by Hirvasniemi et al. Its contribution to pathogenesis of northern epilepsy may not exceed 40%.
Suggesting my evaluations being correct up to here, the conclusion is evident: there must be one or more other genes bearing mutations linked with mutation cln8.001 - surely also with other mutations on CLN8, this will be discussed later on this side - and these genes must besides gene CLN8 be responsible for the documented course of northern epilepsy, predominantly before reaching school age, but also for the period of school age, of puberty and after reaching adult age. Using IMPACD® and comparing expression data for NCL-related genes I came to the following result:
Mutation cln8.001 must generally be linked with a mutation wether on gene NOMO2 or on gene USP10.
Both genes are encoding various proteins belonging to the NCL-genes-and-protein network. As far as I know genes NOMO2 and USP10 (this might be valid for all genes for the first time stated in this work to be linked with NCL in general) were until today never checked for mutations linked with NCL in common or with northern epilepsy or the turkish variant of NCL in particular. Therefore not knowing the disease causing mutation(s) in gene NOMO2 and USP10 in this state of research I may only offer a conjecture in relation to defective proteins coded on these genes, which could be causative for onset and progression of northern epilepsy in connection with mutation cln8.001:
|
Proteins likely affected by a cln8.001-linked mutation on gene NOMO2
|
|
Protein coded on NOMO2 suspected to be defective
|
Coded on gene(s)
|
Period of expression
|
|
PPT isoform 5
|
NOMO2-CLN1
|
from birth till end of year 5
|
|
MTTDC isoform 6
|
NOMO2-CLN3
|
from birth till begin of puberty
|
|
MTTDC isoform 7
|
NOMO2-CES7
|
from birth till begin of puberty
|
|
Nodal modulator 2
|
NOMO2
|
from birth till ~year 35
|
|
TPP isoform 9
|
NOMO2-CTSD-CLN3
|
from end of y5 till begin of adultness
|
|
MTTDC isoform 13
|
NOMO2-CLN3
|
from end of y5 till ~year 35
|
|
PPT isoform 12
|
NOMO2-CES7-CLN1
|
from begin of puberty till adultness
|
|
In the periods being relevant for onset and progression of northern epilepsy NOMO2 is coding for three isoforms of MTTDC (isoform 6, 7 and 13), each being build from two polypeptide chains, one derived from gene NOMO2 - the other one from gene CLN3 respective CES7. One of the related polypeptidechains encoded on CLN3 most likely will be ENSP00000349586, a polypeptid consisting of 253 residues. It will be used to build MTTDC isoform 6.
The overview above will also give an additional explanation for the increasing of seizures in progress of puberty and the further progress of the disease may find its reason in isoform 16 of PPT, calculated to be be activated with reaching adult age. One of the two polypeptidchains building this isoform of PPT is coded on gene NOMO2, the other on gene CES7.
Onset and progression of northern epilepsy of type cln8.001/NOMO2 will surely also be influenced by CTSD isoform 8 - generated using two chains derived from gene NOMO2 and gene FAM117A - and nodal modulator protein 2, ENSEMBLE protein number ENSP00000331851, synthesis of which will begin with year 5 and will end with begin of puberty.
At this point of discussion I should remark, that - related to northern epilepsy - gene NOMO2 is suspected to bear the pathogenic mutation(s) and not gene CLN3, which I expect to have been thoroughly checked for defects in the related group of finnish patients.
Mutation cln8.001 linked with mutation(s) on USP10.
From USP10 will be generated many different transcripts, coding also different isoforms for all together nine members of the NCL-protein-network (see table). From these transcripts/proteins I postulate the following five proteins with their above listed isoforms to be linked with mutation cln8.001:
|
Proteins likely affected by a cln8.001-linked mutation on gene USP10
|
|
Proteins coded on USP10 suspected to be defective
|
Coded on gene(s)
|
Period of expression
|
|
AR isoform 6
|
USP10-SERPINB11
|
from birth till end of y7
|
|
AR isoform 8
|
USP10-CLN8
|
from birth till reaching adultness
|
|
TPP isoform 5
|
USP10-CLN8
|
from birth till begin of puberty
|
|
PPT isoform 8
|
USP10-ARSG-CLN3
|
from birth till till adult (~y21)
|
|
MTTDC isoform 14
|
USP10-SERPINB11
|
from end of y5 till adult (~y21)
|
|
RI isoform 9
|
USP10-CES7-FAM117A
|
end of puberty till ~y30
|
|
Based on expression data evaluated by computational analysis of relations between genes CLN8 and USP10, respective between CLN8 and NOMO2, the above listed proteins may be the most suspicious candidates for being involved in pathogenesis of northern epilepsy, if we search for reasons for onset after school age and progression after puberty and after reaching adult age.
Half of NCL-patients carrying a mutation on gene CLN8 must be hit by an NCL-related mutation on another gene, most of these up to date unknown for its linkage with NCL.
Studying the phylogenetic tree of NCL-causing genes, the data tables I have established for PTT, AR, RI, TPP, MTTDC and CTSD and studying the transcripts of well known NCL-causing genes from CLN1 through CLN1043 it has high probability, that the wanted gene(s) might be found under the above listed genes NOMO2, GPC6, USP10, DHRS7, CES7 and CLN6.
The following table shows a computer-aided prediction for the potential coupling of (16) known CLN8-mutations (base: third of december, 2010) with genetic defects on other genes. The nomenclature follows NCL-mutation database.
|
Mutation
|
Country of origin
|
coupled with defect/
mutation on gene
|
|
cln8.001
|
Finland
|
group A: NOMO2
group B: USP10
|
|
cln8.012
|
Finland
|
GPC6
|
|
cln8.012
|
Pakistan
|
DHRS7
|
|
cln8.010
|
Pakistan
|
GPC6
|
|
cln8.006
|
Pakistan
|
GPC6
|
|
cln8.006
|
Turkey
|
USP10
|
|
cln8.016
|
Turkey
|
NOMO2
|
|
cln8.007
|
Turkey
|
NOMO2
|
|
cln8.005
|
Turkey
|
DHRS7
|
|
cln8.004
|
Turkey
|
DHRS7
|
|
cln8.014
|
Turkey
|
USP10
|
|
cln8.010
|
Turkey
|
CLN6
|
|
cln8.010
|
Italy
|
CES7
|
|
cln8.008
|
Italy
|
GPC6 or DHRS7
|
|
cln8.009
|
Italy
|
NOMO2
|
|
cln8.015
|
Italy
|
NOMO2
|
|
cln8.011
|
Italy
|
USP10
|
|
cln8.017
|
Germany
|
GPC6
|
|
cln8.013
|
Israel
|
GPC6
|
|
This table also should give a good explanation for the observations made by Ranta et al. when studying vLINCL occurring in a group of turkish families. We remember:
Ranta et al. reported homozygosity over the Northern epilepsy CLN8 gene region on 8p23 in four out of five Turkish vLINCL families studied. However, no common mutation in CLN8 was found in these families. Later they extended the Turkish vLINCL family panel to 18 families, of which only one was nonconsanguineous. Nine families were excluded from CLN8 by lack of homozygosity. In the remaining families, four CLN8 gene mutations were identified indicating that in a subset of patients with Turkish vLINCL the disorder is allelic to Northern epilepsy. But there was no apparent genotype-phenotype correlation among the Turkish patients with CLN8 mutations, although their phenotype was distinct from that of Finnish Northern epilepsy patients.
Nine turkish families showed symptomatology of vLINCL, but were excluded from further explorations by lack of homozygosity. In relation to this nine families, I would plead for looking first for mutations on genes DHRS7 and USP10, in case of being unsuccessful for checking NOMO2 or one of the other above mentioned genes too. Same statement is valid for the group of patients with missing genotype/phenotype correlation: the missing phenotype-genotype relation bewailed by Ranta et al. will find its evident cause in an heriditary coupling of mutations on gene CLN8 wether with those on gene NOMO2, USP10, DHRS7 or on CLN6, which I assume to be resident in distinct subsets of the turkish population. Checking the above listed genes for NCL-related mutations should help to find the reason for this phenomen.
CLN6 or CLN8 ?
Linkage of mutation cln8.010 with mutation(s) on CLN6.
Cited from OMIM: Siintola et al. (2005) identified 2 different mutations in the CLN6 gene (606725.0008; 606725.0009) in affected members of 2 Turkish families with CLN6. The findings indicated that a subset of patients with the so-called 'Turkish variant' of late-infantile NCL (CLN7; see 600143), actually have CLN6.
CLN7 in this citation from OMIM must be actually CLN8, to which link 600143 in fact is rigthly set.
I postulate the homozygous 663C-G transversion in exon 6 of the CLN6 gene, resulting in a tyr221-to-ter (Y221X) substitution (606725.0008) in most of cases to be linked with mutation cln8.010 on gene CLN8.
Summary for CLN8
- Mutations on CLN8 in most cases must be linked with at least one mutation in one of genes NOMO2, GPC6, DHRS7, CES7, CLN6 or USP10.
- Mutation cln8.001 should always be linked either with a mutation on NOMO2 (ca. 75% - 17 from 22 cases documented by Hirvasniemi et al. ) or with a genetic defect on gene USP10 (ca. 25% - 5 of 22 cases)
- Mutation cln8.010 in most of cases must be linked with mutation cln6.023 (606725.0008 in OMIM) in CLN6 gene. This statement refers to the turkish population only.
|
CLN9 disease as described by Schulz et al. (2004) must be crucially linked with mutations on gene NOMO2, USP10 or CES7.
CLN9 is defined as a phenotype only of a variant of juvenile NCL, first described and characterised in the case of two Serbian sisters and two German brothers by Schulz et al. (2004). The sisters, whose great-grandmothers came from adjacent villages, developed declining vision, progressive ataxia and seizures at the age of 4 years. At the age of 10 years, they could not walk independently and became mute. The 2 brothers showed a similar course as the sisters, loosing vision and getting seizures at the age of 4 years. Cognitive decline was apparent at an age of 6 years, ataxia and rigidity occurred at an age of 9 years, and they were mute at the age of 12 years.
The younger brother died at age 15 years following pneumonia - the older brother, who later had hallucinations and dysphagia, died at age 19 years.
Schulz et al. later (2006) found that fibroblasts derived from patients with CLN9 showed markedly decreased levels of dihydroceramide and decreased dihydroceramide synthase activity. The cells showed partial correction of growth defects and apoptosis when transfected with CLN8 (607837) and several human LASS genes (see, e.g., LASS1; 606919), all of which increase dihydroceramide synthase activity. Schulz et al. therefore concluded that the CLN9 protein may be a regulator of dihydroceramide synthase.
NOMO2 is located on chromosom 16, short arm, region 12.3 - a region which is well known for gene duplications (see also genetic tree of NCL-causing genes). It is spanning at least 32 exons and is assumed to code two isoforms of the same protein, the so called nodal modulator 2. Assigning a function to this protein seems to be difficult. Cited from NCBI refSeq10:
This gene encodes a protein originally thought to be related to the collagenase gene family. This gene is one of three highly similar genes in a region of duplication located on the p arm of chromosome 16. These three genes encode closely related proteins that may have the same function. The protein encoded by one of these genes has been identified as part of a protein complex that participates in the Nodal signaling pathway during vertebrate development. Mutations in ABCC6, which is located nearby, rather than mutations in this gene are associated with pseudoxanthoma elasticum (PXE). Two transcripts encoding different isoforms have been described. (end of cit.)
I propose the so called CLN9 disease to have its basic cause in mutations in genes NOMO2, USP10 and CES7 - defects on NOMO2 and/or CES7 being likely related to the case of the mentioned german brothers - and in mutation(s) in gene USP10 and/or CES7, the latter most likely connected with the fate of the two serbian sisters, both medical records described by Schulz et al. (see above).
Looking at the expression-tables below, we will realise multiple connections on the molecular level between USP10, CES7 and NOMO2 on one side and genes known for NCL related mutations like CLN1, CLN3, CLN6 and CLN8 on the other side. Furthermore USP10, NOMO2 and CES7 encode well known NCL-causing proteins like PPT, TTP and CTSD as well as some proteins newly identified and postulated to cause NCL by the author of this site.
Looking at these tables, the data tables and the analysis of CLN3 gene below it should be evident once more, that the only way to identify a specific variant of NCL is to identify its genetic base and to evaluate the pathogenic effects of the underlying mutation on the different transcripts generated from the specific defective gene.
According to my analysis of NOMO2 with IMPACD® this gene is encoding 26 different polypeptides during ontogenesis, defined by 6 promotors and 7 poly-A-signals/terminators spread over the gene itself and adjacent 5 and 3 untranslated regions within appropriate subdomain (example for definition of a subdomain see here). 24 of these transcripts I could associate with 8 different NCL-related proteins as shown in the following table (2 transcripts likely not NCL-related and missing here):
|
Gene: NOMO2 on Chromosom 16 p12.3
|
|
Protein
|
Transcripts/
mRNAs
|
Expression periods
|
coded on gene(s)
|
|
AR
|
3
|
day 6 till end of embryogen.
day 110 till day 160
day 200 till birth
|
NOMO2 - CES7
NOMO2 (CAC21645.1)- USP10
NOMO2 - CES7
|
|
CLCN6
|
1
|
beg. of pub. till beg. of adultn.
|
NOMO2-CLN2-CES7
|
|
CTSD
|
2
|
day 200 till birth
end of y5 till reach. adultness
|
NOMO2 - CLN6
NOMO2 - FAM117A
|
|
MTTDC
|
4
|
birth till end of y5
birth till begin of puberty
birth till begin of puberty
end of y5 till ~ year 35
|
NOMO2-DHRS7
NOMO2-CLN3 (235aa)
NOMO2-CES7
NOMO2-CLN3
|
|
NOMO2
|
3
|
birth till end of y5
birth till ~ year 35
end of y5 till adultness (~y21)
|
NOMO2 ENSP00000370883
NOMO2 ENSP00000331851
NOMO2 - CLN6 - DHRS7
|
|
PPT
|
4
|
day 160 till day 200
day 200 till adultn. (~y21)
birth till end of y5
beg. of pub. till beg. of adultn.
|
NOMO2-CLN6
NOMO2-CES7
NOMO2-CLN1
NOMO2-CES7-CLN1
|
|
RI
|
4
|
day 160 till day 200
day 200 till birth
from ~y30 on
from ~y30 on
|
NOMO2-CLN3-USP10
NOMO2-CES7-FAM117A
NOMO2-CLN6
NOMO2-USP10
|
|
TPP
|
2
|
end of y5 till beg. of adultness
from begin of adultness on
|
NOMO2-CLN3-CTSD
NOMO2-GPC6
|
|
USP10
|
1
|
end of pub. till beg. of adultn.
|
USP10-CES7-NOMO2
|
|
USP10 is described to code ubiquitin specific peptidase 10 (ubiquitin carboxyl-terminal hydrolase 10 - EC 3.1.2.15), which belongs to the large family of ubiquitin-specific processing enzymes. Ubiquitin specific peptidase 10 is part of the endosomal endocytotic system.
Taken from NCBI RefSeq: Ubiquitin is a highly conserved protein that is covalently linked to other proteins to regulate their function and degradation. This gene encodes a member of the ubiquitin-specific protease family of cysteine proteases. The enzyme specifically cleaves ubiquitin from ubiquitin-conjugated protein substrates. The protein is found in the nucleus and cytoplasm. It functions as a co-factor of the DNA-bound androgen receptor complex, and is inhibited by a protein in the Ras-GTPase pathway. The human genome contains several pseudogenes similar to this gene. (end of cit.)
Based on analysis of gene USP10 with IMPACD® from this gene during ontogenesis will be synthesised 24 different transcripts, 2 of them likely not NCL-related. From the remaining 22 transcripts most likely 3 are representing isoforms or precursors of ubiquitin specific peptidase 10 (USP10), 19 transcripts are linked with 8 different proteins belonging to the NCL-family too.
|
Gene: USP10 on Chromosom 16q24.1
|
|
Protein
|
Transcripts/
mRNAs
|
Expression period(s)
|
coded on gene(s)
|
|
AR
|
3
|
day 110 till day 160
from birth till end of y7
birth till reach. adultness
|
USP10 - NOMO2
USP10 - SERPIN B11
USP10-CLN8
|
|
CLCN7
|
2
|
during 2nd year
begin of puberty till adult age
|
USP10-FAM117A
USP10 - CLN8
|
|
GPC6
|
1
|
birth till end of y5
|
USP10-FAM117A
|
|
MFSD8
|
1
|
end of y5 - begin of puberty
|
USP10-CLN5-CLN8
|
|
MTTDC
|
2
|
day 160 till day 200
end of y5 till adult age (~21)
|
USP10-DHRS7-CES7
USP10-SERPINB11
|
|
PPT
|
3
|
day 200 till birth
birth till begin of adultness
during puberty
|
USP10-CLN8
USP10-ARSG-CLN3
USP10-FAM117A
|
|
RI
|
5
|
day 160 till day 200
birth till end of y5
end of y5 till begin of puberty
end of puberty till ~y30
from ~y30 on till end of life
|
USP10-CLN3-NOMO2
USP10-CLN3
USP10-FAM117A-CLN1
USP10-CES7-FAM117A
USP10-NOMO2
|
|
TPP
|
2
|
from birth till begin of puberty
birth till begin of puberty
|
USP10-CLN8
USP10-CLN8
|
|
USP10
|
3
|
from day 200 till end of life
begin of puberty till adult age
end of puberty till adult age
|
USP10 ENST00000219473
USP10 ENST00000397953
USP10-CES7-NOMO2
|
|
From gene CES7 analysed with IMPACD® during ontogenesis will be synthesised 33 different transcripts, from these transcripts most likely 5 are representing isoforms of carboxyl esterase 7 (CES7), the remaining mRNAs are linked with 13 different protein-families belonging to the NCL-gene-and-protein-network as listed above.
|
Gene: CES7 on Chromosom 16q12.2
|
|
Protein
|
Transcripts/
mRNAs
|
Expression period(s)
|
coded on gene(s)
|
|
AR
|
2
|
day 6 till end embryogenesis
day 200 till birth
|
CES7-NOMO2
CES7-NOMO2
|
|
CES7
|
5
|
end of y5 - adult (~y21)
end of y5 - begin of puberty
end of puberty - adult (~y21)
birth begin of puberty
begin of puberty-end of life
|
ENSP00000324271
ENSP00000290567
ENSP00000428864
ENSP00000428571
ENSP00000428887
|
|
CLCN6
|
1
|
beg. of pub. till beg. of adultn.
|
CE7-NOMO2-CLN2
|
|
CLCN7
|
1
|
during puberty
|
CES7 - CLN8
|
|
CTSD
|
1
|
end of y5 - end of life
|
CES7-CTSD
|
|
CTSL1
|
3
|
birth begin of puberty
begin of pubertyadult (~y21)
begin of pubertyadult (~y21)
|
CES7-CLN8-GPC6
CES7-CLN8
CES7-CLN8-DHRS7
|
|
CTSL2
|
1
|
???
|
CES7-CLN3
|
|
MFSD8
|
1
|
end of puberty - adult (~y21)
|
CES7-DHRS7-CLN8
|
|
MTTDC
|
4
|
day 160 till day 200
day 200 till birth
birth till begin of puberty
birth till end of puberty
|
CES7-USP10-DHRS7
CES7-CLN3
CES7-NOMO2
CES7-CLN3-GPC6
|
|
PDI
|
2
|
end of y5 till adult (~y21)
end of y5 till adult (~y21)
|
CES7 - CLN5
CES7 - CLN1
|
|
PPT
|
5
|
day 110 till birth
day 200 till adultness
end of y5 till adultness
beg. of pub. till beg. of adultn.
from adult till end of life
|
CES7-CLN1-CLN3
CES7-NOMO2
CES7-CLN8
CES7-NOMO2-CLN1
CES7-CLN2
|
|
RI
|
4
|
day 200 till birth
birth till end of y5
end of y5 till beg. of adultness
end of puberty till ~y30
|
CES7-NOMO2-FAM117A
CES7-CLN3 - DHRS7
CES7-CLN3
CES7-USP10-FAM117A
|
|
TPP
|
2
|
birth till end of puberty
during puberty
|
CES7-CLN8
CES7-CLN3
|
|
USP10
|
1
|
from end of puberty till adult
|
CES7-NOMO2-USP10
|
|
Depending from actual mutation each of the many polypeptides encoded by USP10, CES7 and NOMO2 could be defective or missing - and combinations of mutations on two different genes could occur too.
Therefore it may be nonscientific to list proteins, assumed to cause CLN9. Nevertheless I would postulate - comparing the expression data for NOMO2, USP10 and CES7 with case studies of CLN9 - the following proteins with different isoforms to be the main candidates to cause CLN9 as characterised at the beginning of the chapter (in alphabetical order):
Knowing the distinct mutations on these three genes I would surely be able to make more precise predictions.
Taking all the facts together I am strongly convinced, that the main causes for the above described variant(s) of NCL (designated as CLN9) have to be sought on genes NOMO2, USP10 and CES7 - the CLN9 causing proteins should be mainly missing or defective isoforms of proteins as listed above.
Decreased levels of dihydroceramide and decreased dihydroceramide synthase activity observed by Schulz et al. (2004) most likely will be traceable to missing or defective isoforms of CTSL1, CTSL2, CTSD, GPC6 and/or MTTDC.
CLN10-disease believed to be caused by cathepsin D deficency may not be explainable by a single missing or defective protein.
I postulate gene CLN10/CTSD to encode 16 transcripts - 9 of them coding for proteins belonging to the NCL-protein network (sorted by begin of expression):
|
Expression patterns of NCL-related genes encoded by gene CTSD/CLN10
|
|
Protein coded on CTSD suspected to be defect.
|
Coded on gene(s)
|
Period of expression
|
|
AR isoform 4
|
CTSD - CTSL1
|
from day 160 till day 200
|
|
CLCN7 isoform
|
CTSD - SERPINB11
|
from day 160 till reaching adultness
|
|
TPP isoform 3
|
CTSD - CLN3
|
from day 200 till birth
|
|
CTSD isoform 3*
|
CTSD - DHRS7
|
from day 200 till end of year 5
|
|
CTSD isoform 4*
|
CTSD - DHRS7
|
from day 200 till end of year 5
|
|
CTSD isoform 5
|
CTSD ENSP00000236671
|
from birth till reaching adultness
|
|
TPP isoform 9
|
CTSD - CLN3- NOMO2
|
from end of y5 till begin of adultness
|
|
CTSD isoform 7
|
CTSD ENSP00000384947
|
from end of y5 till reaching adultness
|
|
CTSD isoform 9
|
CTSD - CES7
|
from end of year 5 till end of life
|
|
* different transcripts expressed in different cells
7 other transcripts most likely not NCL related
|
|
Cathepsin D is suspected to cause different phenotypes of congenital NCL and NCL of early childhood40 or school age.
But even if we include in our considerations the different isoforms of cathepsin D coded partially or in total on CLN10/CTSD gene as above documented CTSD may not be the solely cause for the reported cases of CLN10-disease. According to my data CLCN7 protein as well as TPP isoforms 3 and 9 must be involved in this disease too, the grade of involvement depending from patients specific mutation.
According to my evaluations acetoin racemase isoform 4, its two chains assumed to be coded on CTSD and CTSL, will be active from around day 160 until day 200 (end of fetogenesis); one chlorid-ion channel 7 isoform, whose two chains I assume to be encoded on genes CTSD and SERPINB11, will be active from day 160 until reaching adultness; TPP isoform 3, its two chains calculated to be coded on genes CTSD and CLN3, will be active from around day 200 until birth. These three proteins I therefore postulate beneath isoforms 3 and 4 of CTSD to be the main cause for congenital NCL and in particular for microcephalie observed in connection with this disease, so far a mutation(s) on gene CTSD/CLN10 has been verified.
Discussing CLN1, CLN6 and other NCLs I have lost some words about function of acetoin racemase in neurons, cells of oligodendroglia and other cells of the CNS (cited from own work):
[...] acetoin racemase is according to my calculations predominantly expressed in neurons, in cells of oligodendroglia (please read this lines), muscle cells (smooth and striated), lymphocytes, specialised cells of the retina (amacrine-cells and Müller-cells) and in many celltypes of liver and kidneys.
[...] Discussing CLN1 I have postulated acetoin racemase to be part of synaptic bodies and to be resident in membranes of synaptic vesicles.
[...] My own calculations and evaluations based on analysis of concerted expression of distinct gene groups let me postulate, that besides PPT also MTTDC, AR, TPP, CTSL1 and CLCN7 have to be found in synaptic bodies as well as in membranes of synaptic vesicles - synaptic bodies representing a kind of support base for the latter. Additionally to these 6 proteins postulated to be found in synaptic vesicles and -bodies most likely there will be detected CTSL2 and CTSF in synaptic bodies too. (end of citation from own work)
Especially the postulated presence of acetoin racemase in endosomes/lysosomes, in cells of oligodendroglia and in synaptic bodies as well as in membranes of synaptic vesicles in neurons may be coupled with mental retardation or microcephalie observed in connection with CLN10.
Though - according to my evaluations - from gene CLN10 will be generated 5 transcripts for cathepsin D, two of them representing a functional isoform42,
I nevertheless estimate the contribution of defective cathepsin D proteins to pathogenesis of different CLN10-variants in general at not more than 50%. The other 50% must be allocated to the before mentioned proteins AR, CLCN7 and TPP.
Analysis of genelocus CLN3 and discussing function of CLN3P
in human cells.
Preliminary remarks:
Readers, which have had the energy to trudge through this work up to here ;-), will not be surprised me postulating CLN3 like most of NCLs to be a multifactorial and multigenetic disease too - gene CLN3 doubtlessly being a part of the NCL-genes-and-protein network as well as all other genes reviewed on this side. Though gene CLN3 is generating besides 12 transcripts for isoforms of multivalent terpene and terpenoid decyclase (MTTDC) various transcripts for altogether 8 different proteins - beneath those some well known and previously discussed like acetoin racemase, CTSD, CLCN7, PPT, TPP and others (see following table) - I will focus in this part of my work on function of CLN3P, which is mainly suspected to cause CLN3/JNCL. This protein of 438aa I postulate to be a multifunctional transmembrane enzyme cleaving carbon rings of distinct lipids, so of phospho- and sphingolipids and of distinct terpenes and terpenoids (to which most vitamines are belonging). Its various isoforms coded partially or in full on gene CLN3 are also involved in lysosomal functions, may those be linked with a membranous residence or such in the inner lysosome or endosome.
CLN3P is believed to be the solely cause for juvenile NCL (Batten-disease, NCL of Spielmeyer-Sjögren type), what according to the results of my research may be wrong: in relation to CLN3 coupled with common 1 kb deletion contribution of CLN3P to pathogenesis of CLN3/JNCL may not exceed 60%, if we relate it to the whole group of CLN3-mutations this value may be around 50%.
Genelocus CLN3 is believed to cause juvenile NCL (JNCL). Two proteins with identic sequence of aminoacid residues (438aa), but transcripts different in 5 untranslated region have been identified. At least seven more transcripts (base january 2011) are annotated by ENSEMBLE.
Cited from NCBI: This gene encodes a protein that is involved in lysosomal function. Mutations in this, as well as other neuronal ceroid-lipofuscinosis (CLN) genes, cause neurodegenerative diseases commonly known as Batten disease or collectively known as neuronal ceroid lipofuscinoses (NCLs). Many alternatively spliced transcript variants have been found for this gene. (end of cit.)
Though CLN3-gene has been for more than 20 years the the aim of great and expensive efforts to reveal its relevance for Juvenile NCL (in the USA mostly named Batten disease) one was despite of all efforts not successful in identifying the protein (or the proteins) encoded by gene CLN3 and thought to be mainly involved in emergence of one of the most fatal and most widespread forms of NCL - the juvenile form.
According to my data CLN3 gene - which counts in total 15 exons - is equipped with 8 promotors and 5 poly-A-signals (terminators), being combinated to form 35 different transcripts during ontogenesis. These 35 transcripts are representing wether isoforms or precursors / polypeptchains belonging to 9 different distinct NCL-related protein-families as listed below.
Most transcripts encoded by gene CLN3 belong to isoforms of a protein, which the author of this paper postulates to act as a multivalent terpene-and-terpenoid decyclase (MTTDC), an enzyme which he believes to specifically cleave carbon rings of distinct molecules as mentioned before.
Besides coding for two transcript variants of CLN3P - the 438 residues protein - gene CLN3 is encoding two other isoforms of MTTDC (marked red) and 8 precursors/polypeptidchains for building isoforms of this protein. Other proteins being coded on CLN3 represent precursors or single polypeptidchains of well known or still unknown NCL-causing proteins, which need to be cleaved or need to be linked with a second or third chain to become a fully functional metabolic protein too. In detail CLN3 is according to my calculations coding transcripts for the following proteins:.
- acetoin-racemase isoform 2 (CLN3 - DHRS7)
- acetoin-racemase isoform 7 (CLN3 - CTSF)
- acetoin-racemase isoform 11 (CLN3 - CLN8)
- acetoin-racemase isoform 12 (CLN3 - GPC6)
- beta-L-arabinosidase 1 transcript
- cathepsin D isoform 6 (CLN3 - CLN6)
- cathepsin D isoform 10 (CLN3 - CLN6)
- cathepsin L2 isoform (CLN3 CES7)
- chlorid-ion-channel 7 isoform (CLN3-DHRS7)
- multivalent terpene and terpenoid decyclase isoform 1 (CLN3 - DHRS7)
- multivalent terpene and terpenoid decyclase isoform 3 (CLN3 ENSP00000350457 - CES7)
- multivalent terpene and terpenoid decyclase isoform 4 (CLN3 - CLN6)
- multivalent terpene and terpenoid decyclase isoform 6 (CLN3 ENSP00000349586 - NOMO2)
- multivalent terpene and terpenoid decyclase isoform 8 (CLN3 - GPC6 - CES7)
- multivalent terpene and terpenoid decyclase isoform 9 (CLN3- CLN8))
- multivalent terpene and terpenoid decyclase isoform 10 (CLN3 ENSP00000353073)
- multivalent terpene and terpenoid decyclase isoform 11 (CLN3 ENSP00000346650
- multivalent terpene and terpenoid decyclase isoform 12 (CLN3 ENSP00000350523)
- multivalent terpene and terpenoid decyclase isoform 13 (CLN3 - NOMO2)
- multivalent terpene and terpenoid decyclase isoform 15 (CLN3 ENSP00000353116)
- multivalent terpene and terpenoid decyclase isoform 16 (CLN3 - CLN6)
- palmitoyl-protein-thioesterase isoform 1 (CLN3 - CLN1 - CES7)
- palmitoyl-protein-thioesterase isoform 7 (CLN3 - CLN8)
- palmitoyl-protein-thioesterase isoform 8 (CLN3 ENSP00000350457 - USP10 - ARSG)
- retinol-isomerase isoform 2 (CLN3 - NOMO2 - USP10)
- retinol-isomerase isoform 4 (CLN3 - DHRS7 - CES7)
- retinol-isomerase isoform 6 (CLN3 - USP10)
- retinol-isomerase isoform 8 (CLN3 - CES7)
- tripeptidyl-peptidase isoform 1 (CLN3 - CTSF)
- tripeptidyl-peptidase isoform 2 (CLN3 - CLN6)
- tripeptidyl-peptidase isoform 3 (CLN3 - CTSD)
- tripeptidyl-peptidase isoform 8 (CLN3 CLN6)
- tripeptidyl-peptidase isoform 9 (CLN3 - CTSD - NOMO2)
- tripeptidyl-peptidase isoform 10 (CLN3 DHRS7 CLN6 )
- tripeptidyl-peptidase isoform 14 (CLN3 CES7)
|
The impact of 1 kb deletion and other mutations on CLN3 transcripts or
has truncated CLN3P (438aa) a residual catalytic function?
The so called 1 kb deletion on gene CLN3 is the most known and most widespread genetic defect causing NCL. Related to the whole group of NCL-suffering humans around 65% should have this mutation. Based on sufferers on a CLN3-mutation this percentage will increase to circa 80%.
My finding, that CLN3 is not only coding in full four different isoforms of MTTDC, but also many precursors used to build isoforms for this protein by the way offers a convincing explanation for the existence of different variants of JNCL as by example of a newly defined11 variant of juvenile NCL (JNCL) with a protracted course, which is postulated to be caused by a newly detected c.597C>A transversion in exon 8 of CLN3-gene. According to Sarpong, Schottmann et al. the mutation generates a premature termination codon (p.Y199X) truncating the CLN3 protein by ca. 55%. In heterozygous state mutant mRNA transcripts are expressed at the same levels as the wild-type ones, suggesting the absence of nonsense mediated messenger decay. The authors discuss a potential residual catalytic function of the truncated protein as a cause for this mild phenotype of JNCL.
The same explanation may be valid for the observations of Kitzmüller, Haines et al., which stumbled over the paradoxon, that the CLN3-gene on one side is linked with and cause of severe and early-onset NCLs, on the other side mutations in the same gene - most of all the common 1 kb-deletion - are cause of the late onset and milder form of juvenile NCL. To overcome this paradoxon, the authors hypothesise: [..]..that the 1 kb deletion may allow CLN3 residual function. We confirmed the presence of CLN3 transcripts in JNCL patient cells. When RNA silencing was used to deplete these transcripts in cells from JNCL patients, the lysosomes significantly increased in size, confirming the presence of functional protein in these cells. Consistently, overexpression of mutant CLN3 transcript caused lysosomes to decrease in size. We modelled the JNCL mutant transcripts and those corresponding to mouse models for Cln3 in Schizosaccharomyces pombe and confirmed that most transcripts retained significant function as we predicted. Therefore, we concluded that the common mutant CLN3 protein does indeed retain significant function and that JNCL is a mutation-specific disease phenotype.[..]12
While Kitzmüller et al. suppose some CLN3-gene transcripts despite of 1 kb deletion to retain a significant function, Chan, Mitchinson et Pearce believe to be sure, that truncated CLN3 protein will not overcome cell-inherent qualitity controls and therefore will be degraded soon after trancription.13
The here presented data show, that both research-groups may be, depending from the point of view, partially right, because the defective CLN3 gene - though carrying an 1 kb deletion (mutation cln3.001) - is according to my calculations without any doubt generating different functional transcripts (precursors / polypeptides used to build isoforms of MTTDC and of other proteins coded on CLN3), one or more of which could be identic with or similar to the truncated CLN3-protein. With other words: the truncated CLN3P-forms discussed by Kitzmüller et al. on one side and Sarpong, Schottmann et al. on the other side will possibly represent or be similar to functional precursors designed to be combinated with transcripts from other genes of the NCL-Network to form functional proteins and enzymes - by example specific isoforms of MTTDC and AR - and therefore slip through control mechanisms of the related cell. Eigth such precursors should according to my analysis be synthesised from gene CLN3 (see list above) designed to be coupled with precursors from other genes like CES7, NOMO2, DHRS7, CLN8, GPC6 and CLN6.
As before mentioned several times, the impact of not any CLN3 mutation may be restricted to a single transcript or a single isoform of a protein encoded by gene CLN3. The most common mutation by example - cln3.001, the so-called 1 kb deletion - affects not only CLN3P 438aa, but also 5 other proteins being coded on gene CLN3, as there are: beta-L-arabinosidase, CTSD, CTSL2, CLCN7 and - last but not least - PPT.
Onset and progression of CLN3-disease (once more: as for the most, if not all of NCL-diseases too) therefore has to be characterised as an interaction of several distínct proteins - and to determine the contribution of a single protein or single transcript to the course of the disease seems at least for me to be difficult, as long as we do not know, which distinct transcript/protein will be affected during which time and in which distinct cell and what this protein is exactly doing in this cell.
There is also the fact, that likely all genes coding complementary precursors / polypeptidechains for a CLN3-encoded protein - by example for isoforms of CLN3P/MTTDC - are belonging themselves to the NCL-gene-and-protein network - so possibly bearing a NCL-related mutation too.
Though probability of such configuration may be only by ca. 10 % related to all complementary polypeptids, coded on genes listed in table above to be linked on the molecular level with CLN3, we should not quite forget this possibility.
If we expand these reflections on all mutations documented in mutation-databases up to date (49 mutations for CLN3 - january 2011), the list of affected proteins will most likely (I did not check this item) include the whole group of proteins or polypeptides coded on gene CLN3 according to my calculations.
|
Expanding function of CLN3P from a-pinene-oxide decyclase to multivalent terpene- and terpenoid decyclase.
Readers of previous versions of this paper will know, that I first described protein CLN3P 438aa (ENSP00000353073) to be a member of pinene degradation pathway - this according to my data supplied by computational analysis with IMPACD® being the closest linkage to CLN3P I was able to detect in Brenda and Kegg enzyme databases.
Based on expanded calculations, on analysis of reports dealing with wildtype CLN3P and on a self-designed and -programmed computermodel of CLN3P I have come to the conclusion, that human CLN3P must be a multivalent molecular decyclase, whose function is to cleave carbon rings of distinct lipids, so of phospho- and sphingolipids and of distinct terpenes and terpenoids (to which most vitamines are belonging too). Acting as membrane protein in cell walls, nuclear membranes and membranes of most if not all organells in distinct cells it is but also involved in lysosomal functions, may those be linked with a membranous residence or such in the inner lysosome or endosome.
Readers, who are not interested in details about parallels between a-pinene-oxide-decyclase and CLN3P may skip the following chapter and continue here.
A-pinene-oxide decyclase (aPODC) is a black hole in regard of papers describing its function in cell metabolism or in the catalytic pathway of pinene degradation. Alpha-pinene-oxide decyclase is a isomerase, therefore belonging to EC class 5 - but while racemases like acetoin racemase (AR) are acting on hydroxy acids and derivatives, aPODC is thought to do its job as a intramolecular lyase (decyclase) by cleaving both rings of the bicyclic structure of the oxide of a-pinene as it can be seen here. The roles of aPODC respective its putative human ortholog MTTDC shall be investigated more precisely in the following chapters.
aPODC was first purified and characterised by Griffiths et al. in 198715 - them gaining the enzyme from a strain (P18.3) of Nocardia sp.- a gram-posititive bacterium, which was cultured with (+-) a-pinene as sole carbon source. The native enzyme was described by Griffiths et al. to have an apparent molecular weight of 50,000 Dalton by ultracentrifugal analysis. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis gave two dissimilar subunits with apparent molecular weights of 17,000 and 22,000 Dalton. The enzyme was devoid of prosthetic groups, had no cofactor requirement and a broad pH activity range. Griffiths et al. in this paper suppose aPODC to be the enzyme catalyzing the key reaction of a-pinene oxidation in this and organisms like Pseudomonas spp. and others.
 |
Table above taken from: Biotechnologie von Aromastoffen : Transformation von [alpha]-Pinen mit Pseudomonas fluorescens NCIMB 11671 und Trennverfahren mit Zeolithen; Elke Latza; Dissertation : Universität Hannover, Fachbereich Chemie, 1999.
Little is known about aPODC itself, its substrates or its function in other than procariotic organisms like fungi or plants. The following text is taken from: Degradation of Pinene by Bacillus pallidus BR425.16
Pinenes, the major constituents of turpentine, are bicyclic monoterpenes which are produced in significant quantities by plants of the Pinaceae family. Because of their volatility, pinene emissions from conifer forests and during pulping operations constitute a major source of biogenic hydrocarbons (Lindskog&Potter 1995; Stromvall&Petersson 1993). The metabolism of pinenes by microorganisms has been little studied, as they have limited water solubility, and are membrane-destructive to procaryotic and eucaryotic microorganisms (Andrews et al.1980). In an early report in which a number of pinene metabolites were identified, catabolism of alpha-pinene by Pseudomonas strain PL was suggested to proceed by isomerization of the pinene to limonene with subsequent oxidation to perillic acid prior to ring cleavage and further catabolism utilizing a beta-oxidation pathway (Shukla & Bhattacharyya 1968). An alternative pathway through limonene proposed by Gibbons& Pirt (1971) has been questioned (Trudgill 1990). A third pathway and some of the participating enzymes for Nocardia strain P18.3 (Griffiths et al. 1987) and Pseudomonas fluorescens NCIMB 11671 (Best et al. 1987) have been described, in which alpha-pinene is directly oxidized to pinene epoxide prior to ring cleavage. Recent evidence indicates the presence of an alternative pinene pathway in the pinene-epoxidizing Pseudomonas fluorescens strain NCIMB 11671, the metabolites for which have not yet been identified (Colcousi et al. 1996). Few of the enzymes participating in pinene catabolism or their encoding genes have so far been characterized.
Unfortunately until today one was not able to identify the gene (or genes) coding for aPODC even in the strains of Pseudomonas, Nocardia or other bacteria, which were described to use this enzyme as part of pinene degradation as described before.
A complete aPODC catabolism has been verified for Nocardia sp strain P18.3, but also for some Rhodococcus strains19 and different pseudomonas stems. As we have read in the cited abstract from Degradation of Pinene by Bacillus pallidus BR425 there are existing at least three different pathways for pinene catabolism and only few of the enzymes participating in those processes have been identified or characterised until now. But it is generally believed, that cleavage of both rings of the bicyclic monoterpene pinane is needed to get an acyclic substrate for a following beta-oxidation.16
While until now aPODC has been shown to catalyse alpha-pinene degradation only in a few procariotic organisms, degradation of hydrocarbons in general - to which monoterpenes like pinene or limonene and beyond that all terpenoids and terpenes are belonging - has been shown to be a constant property of many bacterias and also many fungi like aspergillus or even yeast.17,18.
aPODC respective its putative multivalent human aequivalent MTTDC until today not being detected or identified in eucariotic cells does of course not mean this enzyme not acting or existing in human cells: an explanation for the lack of data for the postulated MTTDC could be on one side the fact, that one was simply not looking for it, because metabolism of terpenes and terpenoids is studied primarily in procariotic bacteria to get a base for the production of essential oils for the cosmetic-industry, on the other hand it may be difficult or perhaps impossible to detect this enzyme in cultured cells of specific immortalised cell-lines - it not being expressed ubiquitously in human cells, but in distinct cells and moreover in distinct times of ontogenesis as shown in expression data table for MTTDC.20
For better understanding the following remarks it may be necessary to distinguish between MTTDC and CLN3P as following: the terminus CLN3P is used to characterise the specific protein of 438 residues generated from gene CLN3 starting from three different transcripts.
MTTDC in contrast to this definition comprises the whole family of proteins, as listed in respective data table. MTTDC therefore refers to all isoforms postulated to exist from this protein during ontogenesis within many different cells. Those isoforms may be build using transcripts from CLN3, but are often generated from distinct other genes than CLN3 too.
This differentiation should be kept carefully in mind, when studying the following text.
Cell walls of acid-fast bacteria like nocardia have similar properties as membranes of eucariotic lysosomes, of mitochondriae and of other cellular organells.
Nocardia belongs to the actinomycetes, a suborder of actinobacteria - together with bacteria of the genera mycobacterium they have quite unusual cell walls that are waxy and nearly impermeable due to the presence of mycolic acids and large amounts of fatty acids, waxes and complex lipids.21 Mycolic acids are branched hydroxy acids carrying aliphatic chain substituents varying in length. The long-chain aliphatic substituents with 45 and more carbon atoms render the cells acid-fast and resistant against lysozyme and most detergents.
In this regard cell walls of acid-fast bacteria have very much resemblance with membranes of eukariotic lysosomes - membranes, which have to save cells from attacks of acidic and agressive ingredients of these cellular compartments, which deal with digestion and recycling or elimination of cell waste and harmful intruders.
Keeping this in mind and regarding the work of other researchers cited below I am quite sure MTTDC not only to be a part of lysosomal membranes and of the inner lysosome/endosome in distinct human cells I have defined in data tables and will characterise more in detail below, but also to be an essential component of membranes of mitochondriae and of other membranes like those of synaptic vesicles, of ER, of Golgi-apparatus and of cell walls as well as of nuclear membranes of respective cells. As mentioned before, MTTDC should be also present in synaptic bodies (syn.: synaptic ribbons), those organelles being closely related to synaptic vesicles, likely representing a kind of support base for the latter.
That Rakheja et al. found Cln3p residing in lipid rafts or detergent-resistant membranes from bovine brain, in this content may be of special interest as well as the findings of Hobert and Dawson in 2007, who isolated detergent-resistant membranes from control and JNCL brains and found that JNCL-derived membranes were less buoyant than controls. Analysis of phospholipid content showed reduced amounts of bis(monoacyl- glycerol)phosphate (BMP) in JNCL-derived membranes and in total lipids from JNCL brains. Metabolic labeling demonstrated reduced synthesis of BMP also in JNCL fibroblasts, which was restored following complementation with wildtype CLN3P. Hobert and Dawson concluded that CLN3P must play a role in BMP biosynthesis and in maintaining the lipid profile of detergent-resistant membranes in general.
Rusyn et al. found, that CLN3 protein (CLN3p) localizes to Golgi/Rab4-/Rab11-positive endosomes and lipid rafts and postulate it harboring a galactosylceramide (GalCer) lipid raft-binding domain (a postulate, I would agree too). GalCer/mutant CLN3p was retained in Golgi, with CLN3p rescuing GalCer deficits in rafts. Diminishing GalCer in normal cells by using GalCer synthase siRNA negatively affected cell growth/apoptosis of these cells - while adding GalCer restored JNCL cell growth. Wildtype CLN3p was binding GalCer, but not mutant CLN3p. Sphingolipid content of rafts/Golgi was perturbed with diminished GalCer in rafts and accumulation in Golgi. CLN3-deficient raft vesicular structures were small by transmission electron microscopy, reflecting altered sphingolipid composition of rafts. In addition to this they observed CLN1/CLN2/CLN6 proteins were binding to lysophosphatidic acid/sulfatide, CLN6/CLN8 proteins to GalCer, and CLN8 protein to ceramide. Sphingolipid composition resp. morphology of CLN1-/CLN2-/CLN6-/CLN8- and CLN9-deficient rafts were altered.
Rusyn et al. suggest that changes in raft structure and/or lipid stoichiometry could be common themes underlying Ceroid Lipofuscinoses in general (a suggestion, which I estimate to be right only for a minor part of NCLs). And they conclude, that CLN3P impacts galactosylceramide transport, raft morphology, and lipid content33, a conclusion, which according to my evaluations elucidates only a little part of the function of MTTDC/CLN3P, as I will try to show in the following chapters.
Relevance of terpenoids and terpenes for metabolism of eucariotic cells.
Pinene and limonene degradation is part of monoterpenoid metabolism, which is linked with the terpenoid backbone metabolism on one side and on the other side(s) with ß-Alanil metabolism, fatty-acid and lipid metabolism, glucose-alanine cycle (a part of which is the Cori-Cycle) and citrat-cycle. Further connections exist with fatty acid metabolism (a part of which is PPT), with retinol and butanoate metabolism - a part of the former is RI, of the latter AR - with steroid biosynthesis and synthesis of sphingolipids, phospholipids and sphingomyelins. An overview of these parts of global metabolism may be seen here.
Though terpenoids and terpenes are regarded as secondary metabolites only, terpene biosynthesis is one of the most important pathways in synthesis of cellular key substances. All living organisms including humans are using terpene synthesis by coupling units of isoprene (isopentylene). Formally regarded terpenes therefore are oligomers of the hydrocarbon isopren [2-Methyl-1.3-butadien] - that is to say they will be constructed adding isopren-units (C5) to the basic molecule. Depending from the number of C5 isopren-units, terpenes will be classified as monoterpenes (C10 - two units), sesquiterpenes (C15 - 3 units), diterpenes (C20 - 4 units), sesterterpenes (C25), triterpenes (C30) and tetraterpenes (C40). Important cellular key substances and metabolites are derived from terpenes:
Most vitamines and many steroids are belonging to or derived from terpenes or terpenoids: retinol by example is derived from a diterpene, steroids as well as vitamines of the D-group are evolved from triterpenes, while most carotenoids are generated from tetraterpenes.
CLN3P most likely is a multifunctional protein specialised on cleaving carbon rings of specific lipids, in particular of distinct mono-, bi- and polycyclic terpenes and terpenoids.
Protein CLN3P 438aa generated from gene CLN3P, which I have declared to be a isoform of a new putative multivalent terpene and terpenoid decyclase, is - as before mentioned - until now known to be coded by two transcripts, which differ in length of 5 untranslated region. One transcript coding for CLN3P 438aa is declared to have a length of 1913 basepairs, while the other transcript shall count 1827 basepairs45.
CLN3P is believed to be a transmembrane protein presenting 6 putative membrane domains (Janes et al. (1996), which could have - together with several motifs that may undergo post-translational modifications - a function in targeting and anchoring modified CLN3P to distinct biological membranes.22
Storch, Pohl et Braulke reported in 2004 a dileucine motif and a cluster of acidic amino acids in the second cytoplasmic domain of CLN3p (mouse) being required for efficient lysosomal targeting.23 C-terminal prenylation of CLN3p was according to another paper of Storch et al. required for efficient endosomal sorting of CLN3p to lysosomes.24
Based on my above mentioned computer model of CLN3P (438aa - human), I have come to the conclusion, that CLN3P/MTTDC may generally exist in six different functional forms, which in distinct cells and distinct stages of ontogenesis differ in molecular weight due to differing posttranslational modifications and/or polymerisation of the rough endoplasmic reticulum (RER) generated protein.
While the non-modified protein should have a molecular weight of app. 42 kDa (this is appr. equal to the weight of a-pinene-oxide decyclase from P. putida), the posttranslational modified protein according to my calculations weights from ~42 to ~126 kDa, depending from individual modification and polymerisation, which itself in turn is linked with the target of the modified molecule: CLN3P/MTTDC will be synthesized in the RER, modified in the Golgi-apparatus and then - depending from target - integrated as monomer (42 kDa), homodimer (84 kDa) or homotrimer (126 kDa) in cell walls, nuclear membranes, membranes of endosomes/lysosomes, of mitochondriae, of synaptic vesicles and of ER itself. In synaptic bodies (syn.: synaptic ribbons) I have just previously postulated it to be resident too.
The following table is a first try to decipher the code of posttranslational modifications defining the intracellular target of modified CLN3P/MTTDC.
|
Possible posttranslational modifications and localisation of
MTTDC / CLN3-protein 438aa in human cells. 1)
|
|
protein
|
estim. molec. weight (kDa)
|
modifi-
cations defining targeting
|
localisation
of modified protein
|
period of
expression
|
|
CLN3P 438aa
1827bp
ENSP00000353073
|
842)
|
MYR, PAL
|
cell wall
|
from birth till reaching adultness (~y21)
|
|
42, 84, 1262)
|
MYR, FAR
|
nuclear membranes
|
|
lysosom. membranes
of 5 diff. nonneuronal cells, see above
|
|
842)
|
MYR, PAL
|
membranes of mitochondria
|
|
842)
|
MYR, MET, PAL
|
membranes of synaptic vesicles
and in synaptic bodies (s. ribbons)
|
|
842)
|
MYR, PAL, PHO
|
lysosom. membranes
in neurons
|
|
842)
|
MYR, GLY, PAL
|
membranes of Golgi47
|
|
These basic modifications will be surely accompanied by other distinct sorting- or function-defining signals, as by example those described by Storch et al. in the above cited papers.
|
|
FAR = farnesylation
GLY = glycosylation
MET = methylation
MYR = myristoylation
PAL = palmitoylation
PHO = phosphorylation
|
1) based on computer model, the unmodified
protein has a mol. weight of ~42 kDa
2) monomer 50 - 60 kDa (during transport)
homodimer 2 x 42 kDa ~ 84 kDa
homotrimer 3 x 42 kDa ~ 126 kDa
|
|
These modifications will be surely accompanied by other sorting- and trafficking-signals, as by example those before mentioned and described by Storch et al. in 2004 and 2007 (figure A).
Additional to the findings of Storch et al., that CLN3P will be farnesylated on Cys435 I postulate this protein to be myristoylated on Gly280 too, when being transported to lysosomal membranes of microglia cells (2 distinct celltypes, one of them Hortega cells), to satellite oligodendrocytes, oligodendrocytes building myelin-sheats and protoplasmatic astrocytes48. It is acc. to my evaluations the combination of these two modifications - and those others shown in figure A and B - which will lead to trafficking CLN3P to lysosomal membranes of these cells, where they are integrated as homodimers (microglia, protopl. astroc.) or homotrimers (satellite oligodendrocytes, oligodendrocytes building myelin sheats) - corresponding with composition of distinct membrane.
 |
The original of above shown figures (figure B modified by the author of this site) was originally published in JBC - The Journal of Biological Chemistry. Authors: Stephan Storch, Sandra Pohl, Thomas Braulke; A Dileucine Motif and a Cluster of Acidic Amino Acids in the Second Cytoplasmic Domain of the Batten Disease-related CLN3 Protein Are Required for Efficient Lysosomal Targeting;The Journal of Biological Chemistry, December 17, 2004, 279: 53625-53634. © the American Society for Biochemistry and Molecular Biology."
No matter wether this form of CLN3P will be integrated as monomer, homodimer or homotrimer the author of this paper postulates it acting as a membrane protein in distinct previously defined cells and organelles and in this function on one side to be most likely one of the key enzymes in metabolisme of distinct phospho- and sphingolipids and on the other side to be involved in metabolisme of many mono-, bi- and polycyclic terpenes and terpenoids or derivatives of these class of molecules.
Substrates for the different isoforms of MTTDC are therefore in eukariotes resp. in human cells primarily to find amongst distinct phospho- and sphingolipids involved in maintaining function of different cellular membranes and amongst distinct terpenes and terpenoids, some of them belonging to the vitamins.
In total there may exist around 135 different educts/substrates for MTTDC in eukariotic cells - but from those in human cells should be used during ontogenesis not more than 40, most of them belonging to the above mentioned phospholipids and sphingolipids. But triacylgycerides / waxes, glycolipids, steroids, caroteneoids and fatsoluble vitamins will serve as educts / substrates for MTTDC/CLN3P too. Some of these I will present further down.
MTTDC has most likely two different main functions in human cells:
1.
MTTDC is acting as a molecular lyase/decyclase on distinct substances, the structure of which contains one to four carbon rings or one to thirteen isoprene-units.
2.
MTTDC in this function described in 1) is crucially involved in metabolisme or katabolisme of distinct lipids, in particular of phospholipids and sphingolipids (sphingomyelins, ceramids and waxes).
3.
MTTDC in this function described in 1) is involved in metabolisme or catabolisme of distinct terpenes and terpenoids, among these all vitamins belonging to that class of substances or being derived from those molecules.
Involvement of MTTDC/CLN3P in metabolisme of lipids.
MTTDC/CLN3P therefore must be crucially involved in regulating content of most membranous lipids or lipids building myelin sheats of the neuronal axons, as by example phospholipids, glycolipids, sphingomyelins, ceramids and waxes - substances, which are essential for sustaining and regulating homeostasis of yeast vacuoles,25 as well as that of human cellular membranes.
MTTDC/CLN3P must be an essential component of distinct membranes in eukariotic cells, may this be the cell wall, the nuclear membrane, the membrane (myelin sheat) of an axon, the membrane of an lysosome, of the Golgi-apparatus, the ER, of a mitochondrium or that of any lipid-containing organell including membranes of synaptic vesicles. In synaptic bodies (synaptic ribbons) MTTDC/CLN3P should be found too.
There being a strong correlation between degradation of terpenoids and terpenes and degradation of specific lipids, especially sphingo- and phospholipids, the latter being essential components of F0 part of mitochondrial ATP-synthase, function of MTTDS/CLN3P may give besides involvement of TPP, CLCN7 and possibly AR in this process26 also an explanation for the accumulation of F0 subunit c of ATP-synthase, which was reported by Kominami, Ezaki et Wolfe in 1995.27
But most of all accumulation of the so called ceroid lipofuscine - one of the key features in symptomatology of all NCLs - will find an logical and stringent explanation by the function of MTTDC/CLN3P, as described on this side.
In cell walls, in nuclear membranes and membranes of mitochondria, of ER, of Golgi apparatus, in membranes of synaptic vesicles and in synaptic bodies, in membranes of lysosomes / endosomes of all stages and in other biological membranes of a similar composition MTTDC/CLN3P with its various isoforms most likely is part of an inherent membranous recycling and reparation system, which is responsible for sustaining integrity and functionality of these most essential components of all cells.
Correlation between function of CLCN7-protein and MTTDC/CLN3P.
CLN3 gene is (by linkage with gene DHRS7) also encoding a protein of the chlorid-ion channel family, a protein, whose main function may be to regulate pH-value of the inner lysosome and by that activity of enzymes of the inner lysosome, one of which must be MTTDC/CLN3P too.
Most enzymes being present in the inner endosome/lysosome will be active only within a small range of pH. So we should consider - though MTTDC/CLN3P in an actual case possibly not being affected by a mutation and therefore being present in the cell - that its activity could be diminished by a lacking or defective chlorid-ion-channel protein, by example the CLCN7-protein-isoform, whose two chains are encoded on genes CLN3 and DHRS7.
Involvement of MTTDC/CLN3P in metabolisme of vitamins.
It is commonly known, that most vitamins are belonging to the terpenes or terpenoids - or are like the steroids derived from them. Some of those vitamines or prohormones I have just mentioned before.
In addition to previous statements I hereby postulate MTTDC/CLN3P to be crucially involved in metabolisme of distinct steroids and vitamins, which are deduced directly from isoprenoids or terpenes or terpenoids.
Function of MTTDC/CLN3P according to my data therefore should be linked with metabolisme / catabolisme of the following vitamins, resp. prohormones:
Vitamin D3/cholecalciferol, calciol
Vitamin A1 (retinol), Vitamin A (retinal)
β-carotene, α-carotene, γ-carotene, lutein
Vitamin E (α-tocopherol)
Vitamin K1, K2
Vitamins of the B-group, likely B1, B2, B3, B6, B9
Missing or defective isoforms of MTTDC/CLN3P should lead to a severe - possibly time restricted - disturbation in metabolisme of most, if not all vitamins deduced from terpenes, terpenoids or steroids.
From those above mentioned vitamins missing or defective isoforms of MTTDC will likely cause hypovitaminoses of
Vitamin A1 (retinol), Vitamin A (retinal)
Caroteneoids (β-carotene, α-carotene, γ-carotene, lutein)
Vitamin E (α-tocopherol) and
Vitamins of the B-group (likely B1, B2, B3, B6, B9)
and hypervitaminoses of
Vitamin D3 (cholecalciferol, calciol) and
Vitamin K (K1, K2)
Consequences of such vitaminoses may vary, but D3-hypervitaminosis and carotene-, vitamin E- and A/A1-hypovitaminoses will surely be those causing the most severe symptomatology related to CLN3-diseases.
Vitamin E deficiency
In 2002 J.L. Griffin et al.51 report about vitamin E deficiency and related metabolic deficits in an mnd mouse model, which is regarded to be not only a model for motor neuron disease (mnd) but also for CLN8.
The mnd mouse has a profound vitamin E deficiency in sera and brain, associated with the cerebral deterioration characteristic of NCL. In their study, Griffin et al. tried to correct the vitamin E deficiency using dietary supplementation. However, the histopathological features associated with NCL remained. Although vitamin E supplementation reversed some of the metabolic abnormalities, in particular the concentration of phenylalanine in extracts of cerebral tissue, PCA demonstrated that metabolic deficits associated with NCL were greater than any effects produced from vitamin E supplementation. These deficits included increased glutamate and N-acetyl-L-aspartate and decreased creatine and glutamine concentrations in aqueous extracts of the cortex, as well as profound accumulation of lipid in intact cerebral tissue.
Even if these results may not be transferred one to one on conditions in human body the study of griffin et al. gives a strong advice for checking a vitamin E hypovitaminoses in case of CLN3 and perhaps other NCLs too.
Undersupply of vitamin E was also shown to strongly increase formation of lipofuscin in experiments with rats49. Vitamin E deficiency furthermore can cause:
* spinocerebellar ataxia
* myopathies
* peripheral neuropathy
* ataxia
* skeletal myopathy
* retinopathy
* impairment of the immune response
Hypervitaminosis D3
Vitamin D3 in overdose (> 150 µg/l) is highly toxic and causes symptoms, partially similar to those observed in CLN3-disease.
Hypovitaminosis A (Retinol)
The major importance of vitamin A/A1 (retinol/retinal) for biological processes in human body is a well known fact, as well of that of the carotenes.
Hypervitaminosis K
Overdose/surplus of vitamin K has no pathological relevance, nevertheless one should check this item to get an better insight in pathological processes linked with the disease.
Deficiency of vitamins of the B-group
Taken from wikipedia: The B vitamins are a group of water-soluble vitamins that play important roles in cell metabolism. All B vitamins are water-soluble, and are dispersed throughout the body. Most of the B vitamins must be replenished regularly, since any excess is excreted in the urine. The B vitamins may be necessary in order to:
* Support and increase the rate of metabolism
* Maintain healthy skin and muscle tone
* Enhance immune and nervous system function
* Promote cell growth and division, including that of the red blood cells that help prevent
anemia
How to detect a vitamin deficiency?
As mentioned several times before, all proteins being discussed in this paper to cause NCL exist in a more or less great number of isoforms, being activated in different times and mostly different cells too. The chance to detect or verify one or more of the above postulated vitaminoses in case of CLN3-disease/JNCL therefore may be best if we draw our attention to the follwowing time windows:
Vitamin A/Retinol hypovitaminosis should be detectable between end of fetogenesis and end of year 4. During year 5 and 6 levels of retinol in serum may be again normal. From end of year six through to end of year 8 levels of retinol in serum most likely will be zero/naught. From year 9 on levels should again be normal.
The chance to verify a severe vitamin E-hypovitaminosis should be according to my calculations be best from year 5 until year 12/13 (begin of puberty).
Vitamin D3 hypervitaminosis may be observed only from year 5 through to year 10 and from year 12 through to year 15 (during puberty).
A deficiency of vitamins of the B-group may be present starting with year 5 through to year 12/13 (begin of puberty).
Due to specific physiological features in metabolisme of vitamins (building of reserves, storage characteristics) these values are approximate ones. I would suggest to check status of above mentioned vitamins in distinct intervals in combination with regular medical examination of the related patients.
MTTDC/CLN3P most likely being the main cause for vitaminoses observed in connection with CLN3/JNCL one should check occurrence of any vitaminosis also in context with other variants of NCL, especially those, where I have the causing gene(s) declared to encode precursors or isoform(s) of this protein.
All mentioned vitaminoses in case of CLN3-disease may not solely be caused by defective isoforms of MTTDC/CLN3P (though I postulate it being the main cause), but also by defective or missing protein CTSL2 or CTSD respective by a combination of these proteins being defective or missing in distinct time windows and distinct cells.
Correlation between vitamin D3 metabolisme and function of MTTDC/CLN3P.
From those above mentioned vitaminoses I will for a start discuss in this chapter vitamin D3 (cholecalciferol) hypervitaminosis, this disturbation of D3 metabolisme being easily detectable by control of 25(OH)vitamin-D3 level in blood serum of CLN3- or NCL-patients in general.
(This topic is discussed without claiming to have treated it not anywhere near completely!)
Regulation of vitamin D3-metabolism (cited from wikipedia, the free encyclopedia, grammar corrected)
- Cholecalciferol is synthesized in the skin from 7-dehydrocholesterol under the influence of ultraviolet B light. It reaches an equilibrium after several minutes depending on several factors including conditions of sunlight (latitude, season, cloud cover, altitude), age of skin and color of skin.
- Hydroxylation of cholecalciferol to calcifediol (25-hydroxycholecalciferol) by 25-hydroxylase in the endoplasmic reticulum of liver is loosely regulated, if at all, and blood levels of this molecule largely reflect the amount of vitamin D3 produced in the skin or the vitamin D2 or D3 ingested.
- Hydroxylation in the kidneys of calcifediol to calcitriol by 1-alpha-hydroxylase is tightly regulated (stimulated by either parathyroid hormone or hypophosphatemia) and serves as the major control point in production of the most active circulating hormone calcitriol (1,25-dihydroxyvitamin D3).
(end of citation)
Metabolisme of vitamin D3 is a highly complex process, which is up to date not really entirely understood. In particular the regulatory mechanisms to avoid intoxication by high doses of cholecalciferol circulating in blood after exposition to sunlight/UV-radiation are only in part known.
Cholecalciferol is synthesised in the skin from 7-dehydrocholesterol under the influence of ultraviolet B light (sunlight). Cholecalciferol present in serum will be transported - coupled to the vitamin D-binding protein - to the liver, where it will be transformed by Cytochrom P450 to calcidiol (25(OH)vitamin D3). This type of vitamin D3 is the longterm storage form of vitamin D3 in blood - its half-life period averages 19 days.
Due to this half-life-period 25(OH)vitamin D3 is capable to balance irregularities of vitamin D3 production - the level of 25(OH)vitamin D3 in blood therefore mirrors middle- and longterm supply with vitamin D3.
Circulating 25(OH)vitamin D3 will be activated predominantly in the kidneys by hydroxilation to 1,25(OH)2Vitamin D3 (Calcitriol). This process is strictly regulated by different, also very complex mechanisms, which are but not related to the problem discussed in this part of my work.
Cholecalciferol being highly toxic human nature must have developed distinct mechanisms to inactivate a surplus of cholecalciferol circulating in serum due to extensive exposition to sunlight or UV-radiation, one of which is conversion of cholecalciferol to pre-vitamin D3, suprasterol 1, suprasterol 2 or 5,6 trans-vitamin D3 - them all having no hormonic function - under the influence of sunlight or UV-radiation. Conversion of cholecalciferol to tachysterol or lumisterol under same conditions is an additional strategy to eliminate an overdose of highly toxic vitamin D3.
Sunlight under natural conditions not ubiquitously and not permanently present nature must have developed techniques to protect body against cholecalciferol-intoxication by inventing processes, on one side being able to create and sustain an equilibrium between metabolites of vitamin D3 - by example between pre-vitamin D3 and 7-dehydrocholesterol - on the other side being capable to converse a surplus of cholecalciferol in serum to non-toxic intermediates, in case vitamin D3 not being transported quick enough to its storage points mainly in the fatty tissue.
The solution for this problem should be the molecular decyclase MTTDC/CLN3P, capable to do the same as sunlight or UVB-radiation does in relation to the mentioned metabolites of vitamin D3.
That is to say: I postulate MTTDC/CLN3P according to the results of my research to act as molecular sunlight or molecular UVB-light on 7-dehydrocholesterol, pre-vitamin D3, tachysterol, lumisterol and cholecalciferol (vitamin D3), so being in particular an indispensable factor to avoid intoxication with vitamin D3/cholecalciferol and an crucial member of vitamin D3 metabolisme in general.
An compressed insight in vitamin D3 metabolisme gives the following figure C:
 Formation of Vitamin D from 7-dehydrocholesterol in the skin (simplified, according to Holick, M.F. (1995): Environmental factors that influence the cutaneous production of vitamin D. Am J Clin Nutr 61(3 Suppl):638S-645S.) reproduced SVG version of a PNG diagram by Till Reckert, published in wikipedia, the online enzyclopedia. Formation of Vitamin D from 7-dehydrocholesterol in the skin (simplified, according to Holick, M.F. (1995): Environmental factors that influence the cutaneous production of vitamin D. Am J Clin Nutr 61(3 Suppl):638S-645S.) reproduced SVG version of a PNG diagram by Till Reckert, published in wikipedia, the online enzyclopedia.
Related to 7-dehydrocholesterol MTTDC/CLN3P is doing its job by breaking up/eliminating the double bond between C5 and C6 - so opening the B-ring between C9 and C10 of the secosteroid to form previtamin D3 (see figure below).

The more previtamin D3 is present in the dermal cells the less 7-dehydrocholesterol will be synthesised by these cells to avoid a surplus of previtamin D3, which could be rapidly converted to vitamin D3, so possibly leading to toxic doses of D3.
Related to previtamin D3 and vitamin D3 function of MTTDC/CLN3P will likely be linked with breaking up bonding of ring C and D - this statement should be also valid for tachysterol, while lumisterol likely will be cleaved like its isomer 7-dehydrocholesterol. But this and other topics have to be enlighted by further research.
To prove this theory of mine should be easy: control of 25(OH)vitamin D3 levels of CLN3/JNCL-patients - in particular of those with the common 1,0 kb deletion - will without doubt reveal a longterm vitamin D3/cholecalciferol intoxication with levels reaching 200 µg/l on average50, this value representing a severe D3-intoxication. Those levels should be present in all patients of above defined age suffering on the common 1 kb deletion or on any other mutation totally skipping function of MTTDC/CLN3P.
Exceeding formation of tartar/dental calculus in CLN3/JNCL-patients - a sign of D3-hypervitaminosis?
In context with D3-hypervitaminosis an observation made by many parents having actually a child or children suffering on JNCL/CLN3-disease last but not least may support my theory concerning disturbation of D3-metabolisme by lack of MTTDC/CLN3P: they nearly all report heavy problems with tartar/dental calculus for their children, though they all affirm to be more than accurate in dealing with mouth and teeth care for them. I strongly suppose this phenomenon to be linked with D3-hypervitaminosis.
Last but not least: if I had to determine the contribution of above listed vitaminoses to onset and progression of juvenile NCL/CLN3-disease I would set it to 60% in case of 1 kb deletion being the solitary mutation or in case of a mutation with similar consequences, that means resulting in a missing or widely inactive MTTDC/CLN3P. In other cases - in particular if we deal with coupled mutations on CLN3 and USP10 (see next chapter) - this value may be around 40%
These findings open new possibilities and chances to find a causative therapy for JNCL and possibly other forms of this disease within a reasonable time and shed a hopeful light on further efforts to elucidate the real causes of NCLs.
|
Roughly 50% of all NCLs worldwide may not be explainable by a single defective gene: by example 40% of patients suffering on a CLN3 mutation should have at least one NCL-related mutation on gene USP10 too.
As mentioned and documented previously several times, I postulate no distinct NCL-variant to be the result of a single defective protein. Moreover, regarding the phylogenetic evolution of NCL-related genes, it should be obvious, that patients bearing a CLN3 mutation migth also have a mutation in USP10, CES7, DHRS7 or GPC6 being inherited from CLN6, respective CLN3 (GPC6).
In this way USP10 by example may have acquired the common 1 kb deletion, while the other mentioned genes may have inherited only a part of this deletion or another mutation having been present on CLN6 millions of years ago.
Based on calculations, which led to construction of the phylogenetic tree of NCL-related genes, combinations of genetic defects in CLN3 in combination with mutations/defects on other genes should occur as follows:
|
CLN3/USP10
|
40%
|
|
CLN3/DHRS7
|
15%
|
|
CLN3/CES7
|
10%
|
|
CLN3/GPC6
|
5%
|
|
This is to say: 70% of patients bearing any CLN3-mutation should have a genetic defect on a second gene too.
These statement actually focused on CLN3/JNCL has been shown to be valid for other NCLs like CLN1, CLN4 (Kufs disease, adult NCL), CLN5, CLN6, CLN8 and CLN9, which according to my evaluations should also often be caused by a combination of mutated genes, those combinations primarily depending from their relationship to other genes of the phylogenetic tree, which is once more shown on the left side.
In regard to CLN3 (JNCL) the circumstances therefore are similar to those, I above have described for other NCLs like CLN4 (Kufs disease), CLN7, CLN8, CLN9 and CLN10.
From juvenile phenotype of NCL should exist - according to my findings - at least four phenotypes, respective aetiologies caused by particular or combined mutations on distinct genes. These genes are - besides CLN3 GPC6, USP10, DHRS7 and CES7. All these genes and their above listed gene products with varying isoforms and in varying combinations must be involved in onset and progression of different types of juvenile NCL.
According to my findings and my analysis of relations between different NCL-genes, most likely 50% of the whole group of NCL-suffering childs and adults and 70% of patients with a CLN3 mutation suffer on an NCL-related genetic defect on a second gene too, being cause for a great number of partially or totally useless or missing different proteins.
These defects may occur one after another in different stages of ontogenesis, but two or even three being present at the same time depending from hormonal or other activation of involved genes will be observed also.
USP10 deletions correlated with JNCL/CLN3.
Gene USP10 is localized on the forward strand of the long arm of chromosom 16 within band q24.1. The gene spans from bp 84,733,555-84,813,525 and is comprising 15 exons. According to my analysis band 16q24.1 can be divided into 4 chromatin domains separated by REMAKEs (for an example of such structure see here), USP10 is situated within subdomain 1 of chromatin domain 2, which contains 4 subdomains in total.
In fact USP10 should bear many ancestral mutations (and some collected during phylogenesis) and many of these mutations should be closely connected with onset and/or progression of NCLs, specially JNCL.
USP10 1kb deletion aequivalent to CLN3 1 kb deletion.
Using an statistical approach, I guess around 16 mutations existing on USP10 having less or more influence on development and symptomatology of distinct variants of NCL and especially of CLN3/JNCL. One of these mutations related with JNCL (and certainly with other NCLs too) must be an aequivalent of CLN3 1 kb deletion: in USP10 we should find this deletion of around 1000 bp within an intronic region (intron 4-5) beginning likely with bp 84,782,955 and ending likely with bp 84,783,950. This deletion must have without any doubt influence on expression of various distinct transcripts.
This ~1 kb deletion likely being the most widespread NCL-related defect on USP10 the above described mutation should be present in aprox. 40% of patients bearing a genetic defect on CLN3. Related to the whole group of NCL suffering patients therefore some 30% should have at least one additional NCL-related genetic defect on USP10.
Summary and Concluding Remarks.
Associating a distinct protein or enzyme with JNCL or any other NCL would wether be logical nor meaningful.
The documented various mutations in CLN3-gene must affect a great number of precursors and isoforms from up to nine different proteins being generated from gene CLN3 during ontogenesis beginning with day 60 (end of embryogenesis) as for most of these proteins documented in the presented data tables. This may also be the reason for the various differing phenotypes, classical juvenile NCL is presenting itself in different countries or different ethnic groups. Connections on the molecular level between gene CLN3 and many other genes belonging to the NCL-gene-and-protein network - themselves in turn often bearing mutations - makes linkage of classical JNCL to a single specific defective protein even more unlikely and deviously.
According to my computational analysis CLN3-gene is involved in synthesis of 12 from 16 different isoforms of MTTDC being expressed in varying times and different celltypes. As documented in the above presented list, CLN3 besides coding for many isoforms of MTTDC is but also coding polypeptidchains/ precursors used to build isoforms of AR, beta-L-arabinosidase, CTSD, CTSL2, CLCN7, PPT, RI and TPP. The up to date documented 49 mutations on CLN3 (jan. 2011) must without any reasonable doubt affect most of transcripts generated from this gene during ontogenesis. At least 6 different proteins with various isoforms must according to my calculations and evaluations be involved in CLN3-depending variants of NCL.
Therefore I postulate besides multivalent terpene and terpenoid decyclase (MTTDC) to be crucially involved in aetiology of all forms of CLN3/JNCL distinct isoforms of AR, CTSD, CTSL2, CLCN7, PPT, RI and TTP and possibly USP10 (ubiquitin specific peptidase 10). And I postulate these proteins to be together with MTTDC the main cause(s) for the neurological phenomens of the different variants of this disease.
NCL in general must be classified as a multifactorial and multigenetic disease.
All genes being discussed in this paper encode more than one, most of genes more than six different proteins. Depending from specific individual mutation one or more of the encoded proteins will be missing or less or more defective. To link any up to date known variant or phenotype of NCL to a single gene product, a single protein or even a single gene in my eyes therefore seems much to simple.
Taken all my data and conclusions together for me therefore it would be impossible to associate any variant of NCL with a single gene, resp. a single protein or a single isoform of a protein, as it is done until now. Very well knowing this fact possibly being a heavy setback for the untiring efforts to find a therapy for the different phenotypes of NCL, I would do patients as well as other scientists a disservice with not publishing this data. On the other side my evaluations show CLN3 to be the gene, where a therapy could be realised within a short time and would have most likely the most promising effect too.
This statement concerning CLN3-gene may be generally applicable for most if not all NCL-genes - the nomenclature being used today is - citing Alfried Kohlschütter - illogical and based on the historical order of the recognition of NCL-causing genes; at this time no one being aware, that a distinct variant of NCL might not be caused by a single gene or a single gene-product, but by defects on more than one gene and by a combination of defective isoforms of various proteins as stated in this paper.
The data presented on this site show clear and unambiguously NCL not being a monocausal but a multicausal and often a multigenetic disease too, whose different forms or variants arise from a network of genes and proteins, linked on a phylogenetic level as well as on the molecular level. Besides various isoforms of PPT, TTP, CTSD or MFSD8 therefore I am sure different isoforms of MTTDC, acetoin-racemase and retinol-isomerase to be the main cause(s) for the many various variants of NCL.
But protein-disulfide-isomerase, CC-preferring deoxyribonuclease, beta-L-arabinosidase, the cathepsins listed further up and CLCN6 as well as CLCN7 may play a role too, possibly an inferior one, in genesis of the variant forms of NCL and should not be neglected.
With this last version of Aetiology of NCL for a considerable time I strongly believe to have named or identified all NCL related genes and proteins - but this statement is naturally depending from a strict definition of this disease and from marking it off from other similar diseases of the brain and/or the nervous system.
Finally I deeply hope my analysis of the NCL-gene-and-protein network and my findings, that different vitaminoses could be an important contributory cause for CLN3/JNCL and possibly other variants of this disease, as well as the functional characterisation of MTTDC/CLN3P might help to find a cure or a therapy for this and perhaps other variants of NCL.
|
Version 1 published on thursday, 27th of august 2009.
Version 2 published on monday, 23rd of november 2009.
Version 3 published on friday, 4th of december 2009.
Version 4 published on sunday, 30th of january 2011, revised 24th of march 2011.
|

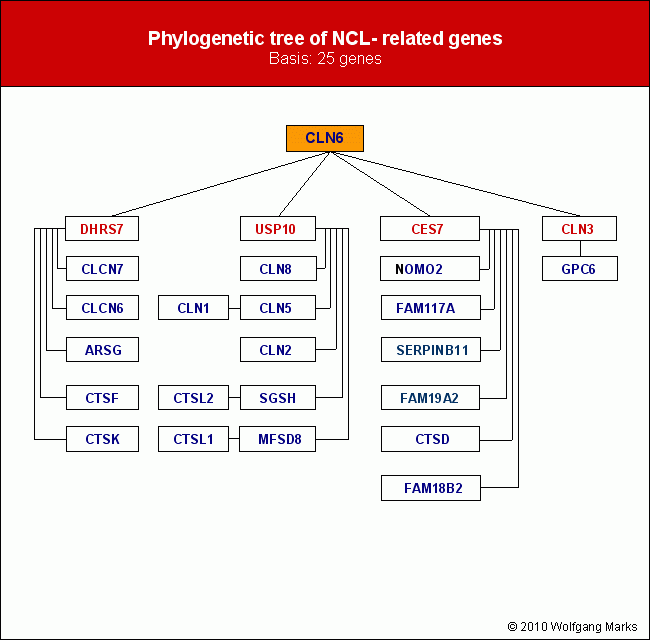

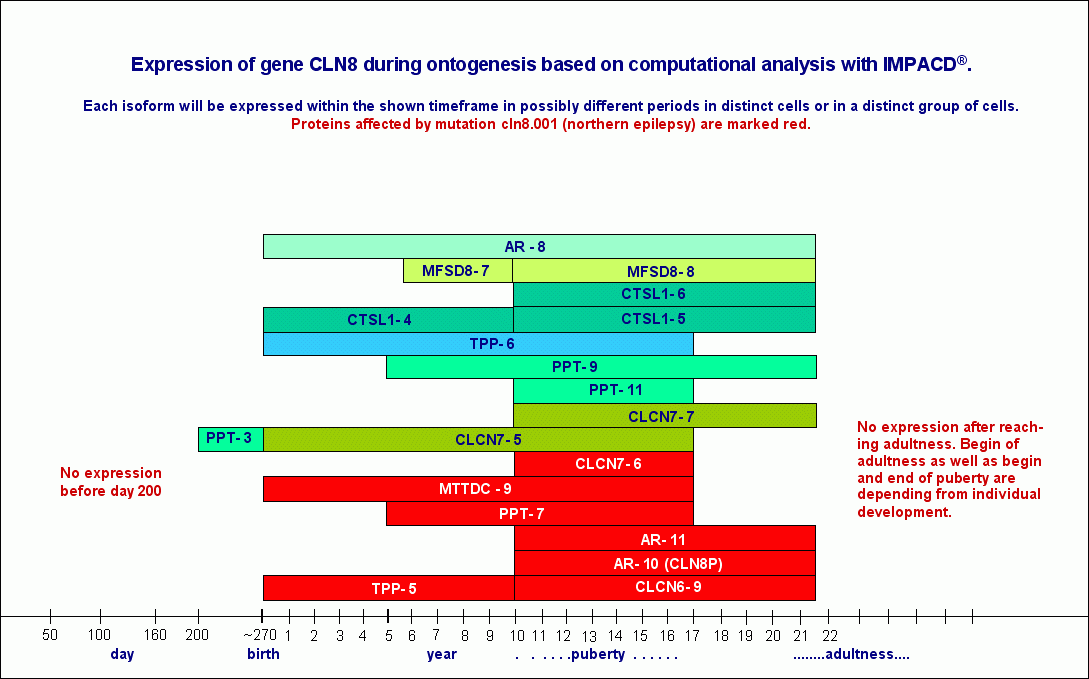



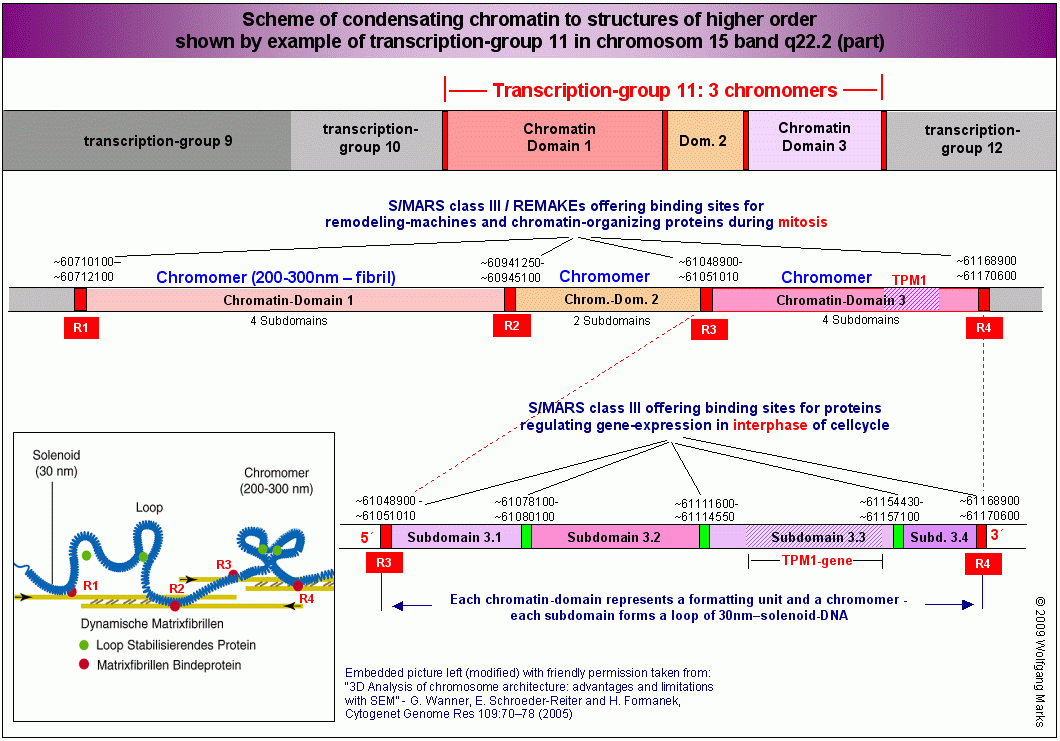{kind=link}